
Receptores acoplados a proteína G (GPCR): qué son y por qué los necesita un tercio de los fármacos
Una proteína que atraviesa la membrana siete veces, cambia de forma al unirse al ligando y enciende una cascada intracelular. Aproximadamente 800 variantes en humanos, blanco del 34% de los fármacos aprobados. Toda la conversación moderna sobre GLP-1, gepants y psicodélicos se juega aquí.
El receptor GLP-1 que ocupa la conversación metabólica de la década es un GPCR. El receptor μ-opioide donde se juega la diferencia entre analgesia y depresión respiratoria es un GPCR. El 5-HT2A que decide si un psicodélico será herramienta antidepresiva o experiencia alucinógena es un GPCR. Entender qué son y cómo señalizan no es lujo académico: es la línea que separa al lector que entiende un Bio de péptidos del que solo se lo cree.
Los dos primeros artículos del Glosario establecieron los ejes que ordenan la conversación farmacológica: qué significa que una molécula sea agonista o antagonista de un receptor, y por qué la vía de administración decide su biodisponibilidad. Pero queda una pregunta que aparece en cada conversación sobre péptidos, GLP-1, opioides o psicodélicos y que no hemos despejado: ¿qué es exactamente un receptor?
Esta pieza responde la pregunta para la familia más importante de receptores en farmacología humana: los receptores acoplados a proteína G, conocidos universalmente por sus siglas en inglés GPCR. Son aproximadamente 800 proteínas distintas en el genoma humano, blanco del 34% de los fármacos aprobados por la FDA, y la diana primaria de prácticamente todos los péptidos terapéuticos relevantes en el catálogo KRECE. El artículo cubre estructura, mecanismo paso a paso, familias de proteína G, la vía β-arrestina, las cinco clases del sistema GRAFS y los GPCRs huérfanos como motor de I+D 2026-2035. Complementa la taxonomía editorial KRECE sobre péptidos al bajar al nivel del receptor diana.
¿Qué es un receptor acoplado a proteína G (GPCR)?
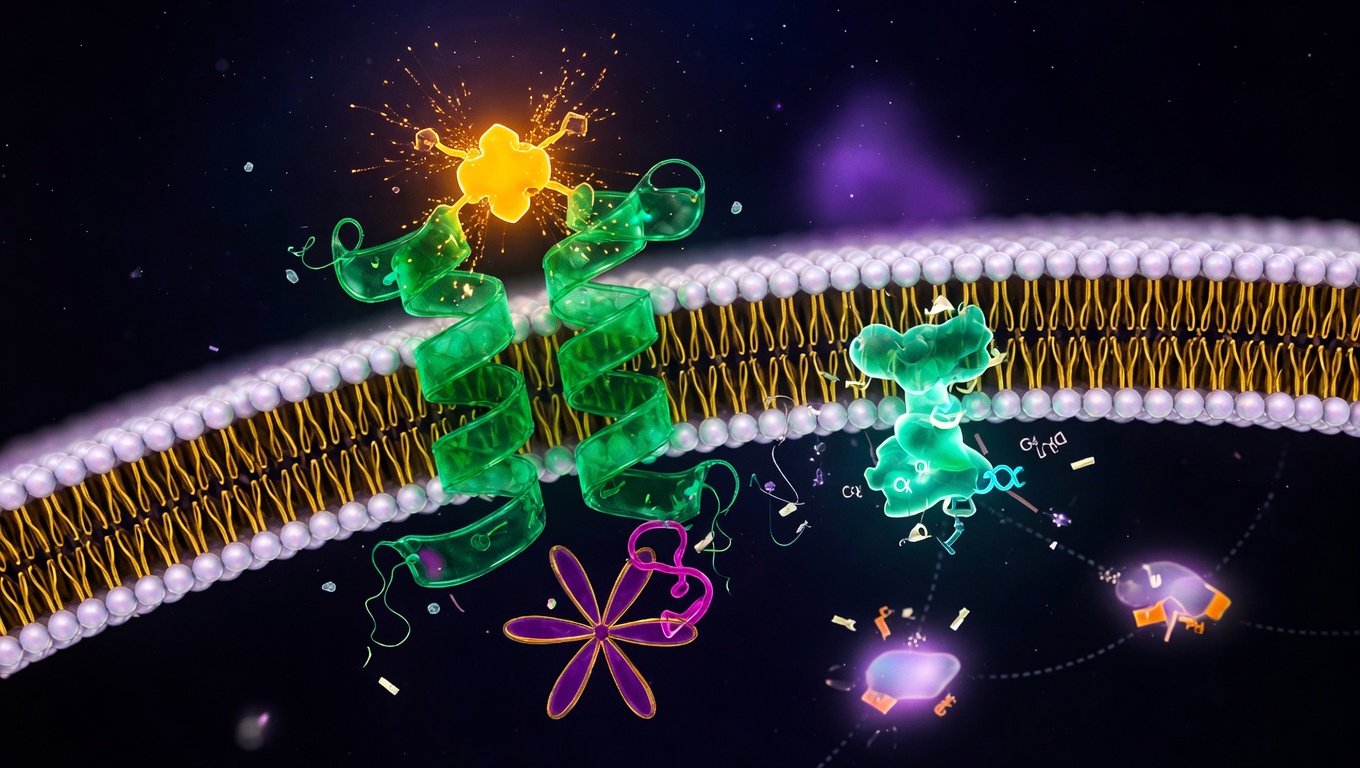
Un receptor acoplado a proteína G es una proteína de membrana que atraviesa la bicapa lipídica siete veces, ofrece un sitio de unión para un ligando en su parte extracelular, y al activarse provoca la disociación de una proteína G acoplada en su parte intracelular, que a su vez dispara una cascada de señalización. La definición parece técnica pero se condensa en una idea sencilla: los GPCR son proteínas-bisagra que convierten una señal química del exterior en una respuesta bioquímica del interior.
Definición técnica: una proteína-bisagra que atraviesa la membrana siete veces
La característica estructural que define a un GPCR es la organización en siete hélices alfa transmembrana. Cada hélice es un segmento de la proteína que cruza la bicapa lipídica de la membrana plasmática de un lado al otro; las siete juntas forman un cilindro de hélices que crea un canal interno donde, en muchos GPCRs, se acomoda el ligando. La nomenclatura técnica habitual es receptor 7TM — siete transmembrana — y es estructuralmente intercambiable con la nomenclatura funcional GPCR.
Por qué se llaman «acoplados a proteína G»
Por la pareja con la que bailan en el lado intracelular. En reposo, el lado citoplasmático de cualquier GPCR canónico tiene asociada una proteína G heterotrimérica — un complejo de tres subunidades llamadas α, β y γ, donde la α sostiene una molécula de GDP. Cuando el ligando activa el receptor, la proteína G se reorganiza: la subunidad α suelta GDP, capta GTP y se separa del par βγ. Ambos fragmentos señalizan por separado hacia distintos efectores intracelulares. Sin la proteína G, la activación del receptor por su ligando no produciría cascada clásica — de ahí el nombre.
Sinónimos y variantes: GPCR, RCPG, receptor 7TM, receptor metabotrópico
GPCR (G protein-coupled receptor) es la nomenclatura anglosajona y la dominante en literatura científica internacional. RCPG es la versión castellana literal — «receptor acoplado a proteína G» — menos usada pero válida. Receptor 7TM es la denominación estructural, útil cuando se quiere enfatizar la arquitectura. Receptor metabotrópico es la denominación funcional, en contraste con receptor ionotrópico: los metabotrópicos actúan indirectamente a través de segundos mensajeros (cAMP, calcio, IP3); los ionotrópicos son canales iónicos que se abren directamente al unirse al ligando (receptores GABA-A, NMDA, AMPA, nicotínicos). Saber distinguir ambos tipos es la base de mucha discusión psiquiátrica — un agonista metabotrópico tiene cinética de segundos a minutos; un ionotrópico cambia el potencial de membrana en milisegundos.
Los números que ordenan la conversación
Cuatro cifras vale la pena memorizar antes de seguir leyendo:
| Cifra | Significado |
|---|---|
| ~800 | GPCRs codificados en el genoma humano (suma de olfatorios y no olfatorios) |
| ~400 | GPCRs no olfatorios funcionalmente caracterizados — la «farmacología real» |
| ~100 | GPCRs huérfanos no olfatorios sin ligando endógeno identificado a 2026 |
| ~34% | Fármacos aprobados por FDA cuya diana es un GPCR (Hauser et al. 2017) |
Las dos primeras cifras explican por qué el genoma humano dedica una proporción tan grande a una sola familia de proteínas: la mitad son receptores olfatorios — la base molecular de nuestro sentido del olfato — y la otra mitad son los GPCRs que median prácticamente toda la comunicación hormonal, paracrina, neurotransmisora y sensorial del organismo. La tercera cifra es la frontera del pipeline farmacológico contemporáneo. La cuarta cifra es la justificación económica de toda la industria farmacéutica moderna sobre receptores.
Anatomía molecular: la estructura siete-transmembrana
Antes del mecanismo, la anatomía. Un GPCR canónico es una sola cadena polipeptídica plegada en siete segmentos helicoidales (TM1 a TM7) que cruzan la membrana, conectados por bucles intra y extracelulares.
Las siete hélices que cruzan la membrana
Cada hélice (transmembrane domain, TM1 a TM7) tiene unos 20-25 aminoácidos de longitud, suficiente para cruzar la bicapa lipídica de unos 30 Å de grosor. Las siete hélices se empaquetan formando un haz cilíndrico con un canal central, en cuyo interior se aloja el sitio de unión del ligando para la mayoría de receptores de moléculas pequeñas (clase A). La organización espacial no es aleatoria: las TM3, TM5, TM6 y TM7 son las que más contribuyen al sitio activo, y la TM6 es la que más se mueve durante la activación — punto fundamental que volverá a aparecer en la próxima sección.
Lado extracelular: donde se une el ligando
El lado extracelular de un GPCR comprende el extremo N-terminal de la proteína (la cabeza polar de fuera) y tres bucles que conectan las hélices TM2-3, TM4-5 y TM6-7 (denominados ECL1, ECL2 y ECL3 por extracellular loop). En los GPCRs de moléculas pequeñas como las monoaminas (dopamina, serotonina, noradrenalina), el ligando entra al canal central del haz de hélices. En los GPCRs de péptidos grandes como el GLP-1R, el N-terminal extracelular es enorme y actúa como una «antena» que captura el péptido antes de que éste alcance la región transmembrana.
Lado intracelular: donde se acopla la proteína G
El lado citoplasmático comprende el extremo C-terminal y tres bucles intracelulares (ICL1, ICL2, ICL3). En el lado intracelular se acopla la proteína G heterotrimérica en estado de reposo, y se acoplan posteriormente las quinasas GRK y la β-arrestina tras la activación y fosforilación del receptor. El extremo C-terminal y el ICL3 son los puntos cláve de fosforilación que disparan la desensibilización — tema central de la sección 05.
Sitios ortostérico vs alostérico
Concepto que el cluster Glosario ya estableció pero conviene reanclar aquí. El sitio ortostérico es la cavidad donde se une el ligando endógeno natural del receptor — el centro del haz de hélices en clase A, el dominio N-terminal en clase B y C. Un agonista o antagonista clásico compite con el ligando endógeno por este sitio. Un sitio alostérico es cualquier otro punto de unión distinto del ortostérico — un bolsillo en la cara intracelular, un hueco entre dos hélices, una interfaz con la membrana. Los moduladores alostéricos positivos (PAM) y negativos (NAM) se unen aquí y modifican la respuesta al ligando natural sin competir directamente con él, lógica de fondo del giro reciente de la industria farmacéutica.
Cómo funciona un GPCR paso a paso
Cinco pasos secuenciales describen el ciclo de un GPCR canónico desde que está en reposo hasta que vuelve a estarlo después de haber señalizado. Es la base molecular de prácticamente toda la farmacología de receptores que aparece en KRECE.
Estado de reposo: el receptor inactivo y el «ionic lock»
En ausencia de ligando, el receptor adopta una conformación cerrada. Dos motivos estructurales mantienen el receptor en estado inactivo. El motivo DRY al final de TM3 (la secuencia conservada Asp-Arg-Tyr, presente en prácticamente toda la clase A) forma un puente iónico con un residuo ácido del extremo intracelular de TM6. Este puente, conocido como ionic lock, mantiene cerrada la interfaz citoplasmática donde se acopla la proteína G. En este estado, el lado intracelular del receptor no puede catalizar el intercambio de GDP por GTP en la proteína G — el sistema está mecánicamente cerrado.
Unión del ligando: el cambio conformacional de TM6
Cuando un agonista entra al sitio ortostérico, su contacto con residuos clave de TM3, TM5 y TM6 induce una serie de reorganizaciones locales que se propagan hacia el lado intracelular. El cambio estructural más importante, documentado por cristalografía del receptor β2-adrenérgico activado (Kobilka et al., Nature 2011), es el desplazamiento hacia afuera del extremo citoplasmático de TM6 en aproximadamente 10-14 Å. Ese movimiento rompe el ionic lock y abre una cavidad citoplasmática donde ahora sí cabe la subunidad α de la proteína G. La transmisión de la señal desde el sitio extracelular del ligando hasta el sitio intracelular de la proteína G es esencialmente mecánica: una cascada de movimientos helicoidales a lo largo del receptor.
Activación de la proteína G heterotrimérica
Con la cavidad citoplasmática abierta, el receptor actúa como un factor de intercambio de nucleótidos de guanina (GEF) para la proteína G. Cataliza la liberación del GDP que la subunidad α tenía unido en reposo, y la entrada de un GTP nuevo (mucho más abundante en la célula). El cambio GDP → GTP modifica la conformación de la subunidad α, que se separa del par βγ. A partir de este punto, las dos fracciones — Gα-GTP y Gβγ — señalizan por separado hacia distintos efectores intracelulares.
Segundos mensajeros: la amplificación de la señal
La fracción Gα-GTP activa o inhibe un efector enzimático de membrana — adenilato ciclasa, fosfolipasa C, RhoGEF, etc., dependiendo de la familia de proteína G (siguiente sección). Ese efector produce un segundo mensajero: una molécula pequeña (cAMP, IP3, DAG) o un ión (calcio) que se difunde por el citoplasma activando kinasas y otras proteínas downstream. La lógica de amplificación es geométrica: un solo receptor activado puede activar muchas proteínas G en un ciclo, cada proteína G genera muchos segundos mensajeros, cada mensajero activa muchas kinasas. Una sola molécula de ligando puede modificar la actividad de cientos o miles de proteínas intracelulares.
Cese de la señal: GTPasa, fosforilación, internalización
Cuatro mecanismos paralelos apagan la señal. Primero, la subunidad Gα hidroliza intrinsecamente su GTP a GDP mediante actividad GTPasa propia, volviendo al estado inactivo y reasociándose con βγ. Segundo, el receptor activado es fosforilado por quinasas GRK (G protein receptor kinases) en residuos serina y treonina del C-terminal y del ICL3. Tercero, esta fosforilación recluta β-arrestina, que desacopla físicamente el receptor de la proteína G. Cuarto, la β-arrestina dirige el receptor hacia endocitosis mediada por clatrina: el receptor se internaliza en endosomas, donde puede ser reciclado de vuelta a membrana o degradado en lisosomas. Esta es la base molecular de la desensibilización y la downregulation en uso crónico de agonistas que ya cubrimos en el primer cornerstone del cluster.
Las cuatro familias de proteína G y qué hace cada una
La identidad de la proteína G acoplada a un GPCR determina qué segundo mensajero se produce y, por tanto, cuál es el efecto fisiológico de la activación del receptor. Hay cuatro familias funcionales basadas en la identidad de la subunidad α.
Gs: enciende adenilato ciclasa → sube cAMP
La s es por stimulatory. La Gαs-GTP activa la enzima de membrana adenilato ciclasa, que convierte ATP en cAMP (AMP cíclico) — uno de los segundos mensajeros más antiguos descubiertos en farmacología (Sutherland, Nobel 1971). El cAMP a su vez activa la proteína kinasa A (PKA), que fosforila docenas de proteínas diana intracelulares. La activación via Gs media efectos clásicos: lipólisis adipocitaria, glucogenólisis hepática, taquicardia. Ejemplos farmacológicos canónicos: receptores β-adrenérgicos (adrenalina), receptores D1 dopaminérgicos, receptores de glucagón, receptores GLP-1R en células beta pancreáticas, receptores GHRH-R en hipófisis.
Gi/Go: apaga adenilato ciclasa → baja cAMP
La i es por inhibitory. La Gαi inhibe la adenilato ciclasa, bajando cAMP y, por extensión, atenuando la actividad de PKA. La subfamilia Go (predominante en neuronas) actúa también sobre canales iónicos: abre canales de potasio (hiperpolariza la célula, la inhibe) y cierra canales de calcio voltaje-dependientes (reduce la liberación de neurotransmisor). Ejemplos farmacológicos: receptores μ-opioide, α2-adrenérgicos, D2 dopaminérgicos, M2 muscarínicos, cannabinoides CB1 y CB2, mayoría de receptores de serotonina excepto 5-HT2 y 5-HT3.
Gq/G11: enciende fosfolipasa C → IP3 + DAG → libera calcio
Gαq activa la fosfolipasa C-β (PLCβ), que hidroliza el fosfolípido de membrana PIP2 generando dos segundos mensajeros simultáneamente: IP3 (inositol trifosfato), que se difunde al retículo endoplásmico y libera calcio del reservorio intracelular; y DAG (diacilglicerol), que activa la proteína kinasa C (PKC). El resultado neto es una elevación abrupta de calcio citoplasmático más activación de PKC. Ejemplos: receptores M1 y M3 muscarínicos (contracción del músculo liso intestinal y vesical, secreciones glandulares), α1-adrenérgicos (vasoconstricción), 5-HT2A (psicodélicos), receptores de histamina H1, vasopresina V1, oxitocina, TRH, GnRH.
G12/G13: regula citoesqueleto y motilidad celular
La última familia y la menos estudiada clínicamente. Gα12 y Gα13 activan factores de intercambio para la GTPasa pequeña Rho (Rho-GEFs), que reorganiza el citoesqueleto de actina. El resultado es regulación de morfología celular, migración y adhesión. Relevancia farmacológica predominantemente en oncología (migración tumoral, metastasis) y desarrollo embrionario. Hasta ahora con pipeline limitado en longevidad pero objeto de investigación activa.
Por qué el mismo ligando puede acoplarse a varias proteínas G
Una sutileza importante. Muchos GPCRs se acoplan promiscuamente a más de una familia de proteína G, con jerarquía relativa dependiente del tejido, del nivel de expresión y del estado celular. El receptor β2-adrenérgico, por ejemplo, se acopla primariamente a Gs pero también a Gi en condiciones específicas. El GLP-1R se acopla primariamente a Gs pero con señal simultánea a Gq en algunas células beta. Esta promiscuidad es precisamente lo que permite el agonismo sesgado — diseñar un ligando que active preferentemente una de las proteínas G sobre las otras, o que active la cascada de la proteína G pero no la de β-arrestina.
La vía β-arrestina: el descubrimiento que rompió el modelo lineal
Durante décadas, el modelo de funcionamiento de los GPCRs fue lineal y unidireccional: ligando → receptor → proteína G → segundo mensajero → efecto clínico. La β-arrestina tenía un papel secundario, como «borradora» de la señal. El trabajo de Robert Lefkowitz y Brian Kobilka revoluciono esta visión y los llevó al Premio Nobel de Química en 2012 — reorientando el campo entero de farmacología de receptores.
Qué es la β-arrestina y por qué Lefkowitz y Kobilka ganaron el Nobel
La β-arrestina es una proteína intracelular soluble que se une a los GPCRs fosforilados en el lado citoplasmático. Hay dos isoformas funcionales en humanos: β-arrestina-1 y β-arrestina-2. Lefkowitz comenzó estudiándolas en los años 80 como mecanismo de «frenado» del receptor β-adrenérgico, y descubrió después, en los años 2000, que tienen función señalizadora propia. Kobilka, su discípulo, cristalizó en 2011 el receptor β2-adrenérgico en estado activo, capturando la imagen molecular que el campo necesitaba para validar todo el modelo. Premiados conjuntamente por la Academia Sueca en 2012.
Función clásica: desensibilización e internalización del receptor
La función original descrita es la de desensibilización homologa. Cuando un receptor está activado y señalizando, las kinasas GRK fosforilan su cola C-terminal en residuos específicos. Esa fosforilación crea sitios de unión para la β-arrestina, que se acopla al receptor y bloquea físicamente la interfaz donde se acoplaría la proteína G. El resultado: el receptor sigue uniendo ligando pero ya no señaliza por la vía G. La β-arrestina recluta también maquinaria de clatrina para internalizar el receptor en endosomas, donde puede ser reciclado o degradado.
Esta vía explica la base molecular de la tolerancia farmacológica en uso crónico de agonistas potentes — los opioides, los β-agonistas en asma, los secretagogos del GHSR como MK-677 e hexarelina. Es la cadena de eventos que el cluster Glosario ha venido refiriendo como downregulation.
Función moderna: cascada de señalización propia, independiente de proteína G
La revelación del campo a partir de 2005-2010 fue que la β-arrestina no es solo un freno; es también una señalizadora independiente. Una vez unida al receptor, actúa como proteína de andamiaje (scaffold) que organiza complejos multiproteícos en el lado intracelular. Estos complejos activan cascadas propias, particularmente la vía MAPK / ERK1/2 (kinasas reguladas por señales extracelulares), con cinética más lenta y persistente que la proteína G y con localización subcelular distinta (frecuentemente desde endosomas internalizados).
El resultado conceptual: un GPCR no señaliza por una vía, señaliza por dos — la vía de proteína G y la vía de β-arrestina — y un mismo ligando puede modular el balance entre ambas. Esto reescribió el diseño farmacológico moderno.
Por qué importa para el agonismo sesgado
Esta es la conexión con el cornerstone Agonistas y Antagonistas del cluster. El agonismo sesgado consiste precisamente en diseñar moléculas que activen un GPCR pero con sesgo hacia una vía u otra: típicamente, se busca un agonista que active la vía de proteína G (asociada al efecto terapéutico) sin activar la vía de β-arrestina (asociada en muchos casos a efectos adversos). La oliceridina como agonista μ-opioide sesgado hacia proteína G frente a β-arrestina — en teoría analésico con menor depresión respiratoria — es el caso paradigmático. La lógica solo es posible porque sabemos desde 2005-2010 que la β-arrestina tiene vía propia, no es un mero apagán.
Las cinco clases de GPCRs (sistema GRAFS)
Fredriksson, Schiöth y colaboradores propusieron en 2003 (Molecular Pharmacology) un sistema de clasificación filogenética que se ha impuesto como estándar: GRAFS es el acrónimo de las cinco familias Glutamate, Rhodopsin, Adhesion, Frizzled/Taste, Secretin. Cada clase agrupa receptores con homología estructural y patrones de unión de ligando similares.
Clase A (rhodopsin-like): la familia más grande, ~85% de los GPCRs
La clase más numerosa y mejor caracterizada. Reúne los receptores de monoaminas (dopamina, serotonina, noradrenalina, adrenalina, histamina), receptores de péptidos pequeños (opioides, oxitocina, vasopresina), receptores muscarínicos de acetilcolina, receptores de quimiocinas, receptores de nucleótidos (adenosina, ATP, ADP), receptores cannabinoides CB1/CB2, receptores de prostaglandinas y leucotrienos, receptores olfatorios y los receptores de la luz (rodopsina). El ligando se acomoda en una cavidad central dentro del haz de hélices. Es la familia diana de la mayoría de fármacos clásicos: antiparkinsonianos (dopaminérgicos), antipsicóticos, antidepresivos (parcialmente), antihistamínicos, opioides, betabloqueantes, broncodilatadores β2, antagonistas H2 gástricos, cafeína, antimigrañosos triptanes y lasmiditán.
Clase B (secretin-like): receptores de péptidos largos
Aproximadamente 15 receptores en humanos pero con relevancia farmacológica desproporcionada. Caracterizados por un extremo N-terminal extracelular grande (~120-160 aminoácidos) que captura péptidos largos (30-50 aminoácidos típicamente) en un mecanismo de unión bipartito: primero la «cabeza» del péptido se une al N-terminal, luego la «cola» del péptido alcanza la región transmembrana y activa el receptor. Reúne los receptores de incretinas (GLP-1R, GIPR), glucagón, secretina, GHRH, paratohormona (PTH), calcitonina, CRH y CGRP. Es la familia diana de los blockbusters metabólicos contemporáneos: semaglutida y tirzepatida en GLP-1R/GIPR, tesamorelina en GHRH-R, los gepants (rimegepant, ubrogepant, atogepant) en CGRP-R para migraña, teriparatida en PTH-R para osteoporosis.
Clase C (glutamate-like): metabotrópicos del SNC y sensor de calcio
~20 receptores caracterizados por un dominio extracelular gigante en forma de Venus flytrap («atrapamoscas») que captura el ligando antes de transmitir el cambio conformacional a la región transmembrana. Reúne los receptores metabotrópicos de glutamato (mGluR1-8), receptores GABA-B (distintos de los GABA-A que son ionotrópicos), el receptor sensor de calcio extracelular (CaSR) y un grupo de receptores del gusto T1R. Relevancia farmacológica creciente: cinacalcet como PAM del CaSR para hiperparatiroidismo, baclofeno como agonista GABA-B en espasticidad, moduladores alostéricos de mGluR2/3 y mGluR5 en pipeline psiquiátrico para depresión resistente y síndrome de X frágil.
Clase F (frizzled/taste): Wnt y receptores de sabor amargo
La más pequeña y atípica. Incluye los receptores Frizzled (FZD1-10) que median la señalización Wnt en desarrollo embrionario, homóstasis epitelial y oncología; el receptor Smoothened (SMO) en la vía Hedgehog; y los receptores de sabor amargo (T2Rs). Relevancia farmacológica creciente en oncología: el vismodegib y sonidegib son inhibidores de Smoothened aprobados para carcinoma basocelular avanzado. Los receptores T2R amargos son objetivo emergente en investigación metabólica por su expresión en células L intestinales (secreción de GLP-1).
Clase Adhesion: la familia con dominio extracelular gigante
~33 receptores en humanos, caracterizados por un dominio N-terminal extracelular extraordinariamente grande con motivos de adhesión celular (GAIN, EGF-like, lectina, etc.). Función fisiológica todavía parcialmente caracterizada en muchos casos; relevancia probada en desarrollo neural, sistema inmune e inmunooncología. El receptor más estudiado es GPR56 (ADGRG1) en desarrollo cortical. Pipeline farmacológico todavía temprano pero con tracción creciente.
GPCRs huérfanos: el pipeline del próximo decenio
Los GPCRs huérfanos son aquellos cuyo gen ha sido identificado en el genoma humano pero cuyo ligando endógeno — la hormona, neurotransmisor o molécula natural que los activa — sigue sin conocerse. A 2026, aproximadamente 100 GPCRs no olfatorios humanos permanecen huérfanos — un cuarto del total funcional. Son la frontera de I+D farmacológica.
El proceso de identificar el ligando se llama deorphanization («deshuerfanización»). Una vez deorphanized, el GPCR pasa de curiosidad genética a diana terapéutica validable. Casos relevantes recientes: GFRAL, receptor del péptido GDF15, deorphanized en 2017 por cuatro grupos independientes (Mullican et al. Nat Med, Yang et al. Nat Med, Hsu et al. Nature, Emmerson et al. Nat Med, todos publicados prácticamente a la vez). GFRAL se expresa exclusivamente en tronco encefálico y media efectos de pérdida de apetito; agonistas en desarrollo clínico para obesidad. GPR75, identificado en 2021 (Akbari et al. Science) como gen cuyas variantes de pérdida de función se asocian a menor índice de masa corporal — potencial blanco terapéutico anti-obesidad de próxima generación. GPR55 en sistema endocannabinoide ampliado, todavía en caracterización.
Los GPCRs que importan en longevidad y péptidos
Catalogo de los receptores donde se juega la conversación clínica de longevidad y péptidos en KRECE. Cada uno con su clase GRAFS, proteína G dominante y fármacos representativos.
GLP-1R: clase B, target de semaglutida, tirzepatida y retatrutida
El GPCR más mediatizado de la década. Receptor de clase B, acoplado primariamente a Gs (eleva cAMP en célula beta pancreática → aumenta secreción de insulina glucosa-dependiente), también a Gq en algunos contextos. Semaglutida es agonista completo (~89% biodisponibilidad SC); tirzepatida es super-agonista dual GLP-1R/GIPR; retatrutida es triple GLP-1R/GIPR/glucagón-R. La técnica de cristalización del GLP-1R activado (Liang et al. Nature 2017) facilitó el diseño de los super-agonistas duales y triples actuales.
GHSR (receptor de ghrelina): clase A, target de MK-677 e ipamorelina
Receptor de clase A acoplado a Gq, con señalización constitutiva basal — lo que lo convierte también en target potencial de agonistas inversos. Ligando endógeno: ghrelina, hormona del hambre producida por estómago. Agonistas peptidícos: ipamorelina, GHRP-6, hexarelina. Agonista no peptidíco oral: MK-677 (ibutamoren). Famosos por desensibilización rápida con uso crónico — ejemplo paradigmático del cluster Glosario sobre la lógica del cycling de péptidos.
GHRH-R: clase B, target de tesamorelina y sermorelina
Receptor de clase B acoplado a Gs en la adenohipófisis. La activación libera GH endógena en pulsos manteniendo los bucles regulatorios negativos — diferencia fundamental con GH exógena. Tesamorelina aprobada para lipodistrofia VIH; sermorelina en uso compounded extendido.
MC4R (melanocortina): clase A, target de PT-141 y setmelanotida
Receptor de clase A acoplado a Gs, expresado en hipotálamo (regulación de apetito y balance energético). Setmelanotida es agonista selectivo MC4R aprobado para obesidad monógena por deficiencia POMC. PT-141 (bremelanotida) agonista MC3/MC4 con efecto sobre vías centrales relacionadas con deseo sexual.
Receptores adrenérgicos: clase A, target de betabloqueantes y broncodilatadores
Familia de nueve subtipos (α1A, α1B, α1D, α2A, α2B, α2C, β1, β2, β3) que median prácticamente toda la señalización simpática. α1 acoplados a Gq (vasoconstricción); α2 a Gi; β1 y β2 a Gs (taquicardia, broncodilatación, lipólisis); β3 a Gs en tejido adiposo. Antagonistas selectivos β1 (bisoprolol, metoprolol) son estándar cardiovascular; agonistas β2 (salbutamol, formoterol) son estándar asma/EPOC.
Receptores opioides μ/κ/δ: clase A, target de morfina, naloxona, oliceridina
Tres subtipos de clase A acoplados a Gi/Go (inhibición de adenilato ciclasa, apertura de canales de potasio, cierre de canales de calcio). Morfina, fentanilo y oxicodona son agonistas μ-opioide clásicos. Naloxona y naltrexona son antagonistas competitivos. Oliceridina (aprobada FDA 2020) es el primer agonista μ-opioide sesgado hacia proteína G en clínica.
Receptores 5-HT2A: clase A, target de pimavanserina y psicodélicos en desarrollo
Receptor serotoninérgico de clase A acoplado a Gq. Diana de los psicodélicos clásicos (psilocibina, LSD, DMT) y de la pimavanserina como agonista inverso para psicosis asociada a Parkinson (aprobada FDA 2016). Pipeline activo en agonismo sesgado para disociar el efecto psicoplástico (potencial antidepresivo) del efecto alucinógeno — uno de los puntos calientes de I+D 2025-2030.
GFRAL/GDF15: receptor deorphanized en 2017 con potencial en obesidad
El caso más reciente de deorphanization con tracción clínica directa. GFRAL es un receptor de clase con dominio Rec extracelular, expresado exclusivamente en núcleo del tracto solitario del tronco encefálico. Su ligando, GDF15 (growth differentiation factor 15), media respuestas de anárexia/náusea ante estrés celular. Agonistas y biológicos en desarrollo clínico fase 1-2 para obesidad, con la hipótesis de que activación GFRAL produce pérdida de peso por un mecanismo no incretina — potencialmente complementario a GLP-1.
Por qué los GPCRs son la frontera de la farmacología actual
Si el 34% de los fármacos aprobados ya actúan sobre GPCRs, ¿por qué la industria sigue invirtiendo enormemente en esta familia? Cuatro vectores técnicos explican el momento.
Agonismo sesgado: el dial fino que separa efecto deseado de efecto adverso
Una vez aceptado que un mismo GPCR señaliza por dos vías paralelas (proteína G y β-arrestina), diseñar ligandos sesgados hacia una vía u otra es el modo más sofisticado de mejorar el cociente terapéutico. Oliceridina (μ-opioide), lasmiditán (5-HT1F), pipeline activo en 5-HT2A para psicodélicos con efecto antidepresivo sin alucinación. Toda la lógica reposa sobre el descubrimiento de la señalización propia de β-arrestina.
Modulación alostérica PAM/NAM: cambiar el receptor sin sustituir al ligando
Como cubrimos en el cornerstone Agonistas, los moduladores alostéricos positivos y negativos actúan en sitios distintos del ortostérico, amplificando o atenuando la respuesta al ligando endógeno sin sustituirlo. Las benzodiacepinas como PAM de GABA-A (clase no GPCR pero misma lógica), cinacalcet como PAM del receptor sensor de calcio (CaSR, clase C), pipeline activo en moduladores alostéricos de mGluR2/3 y mGluR5 para depresión y X frágil. La ventaja técnica clave es el techo de seguridad por preservación del patrón endógeno.
Dimerización y heterómeros: la complicación que reabre el campo
El modelo clásico asume que los GPCRs son monómeros funcionales. La evidencia acumulada en los últimos años indica que muchos GPCRs forman dimeros (con otro GPCR idéntico u homologo) o incluso heterómeros (con un GPCR distinto), y que estos complejos tienen propiedades farmacológicas emergentes — sensibilidad a ligandos distinta, acoplamiento a proteína G distinto, perfil de desensibilización distinto. El campo es técnicamente complejo y todavía no se ha traducido en gran escala a fármacos clínicos, pero abre un espacio de diseño farmacológico nuevo.
GPCRs huérfanos: deorphanization como motor de I+D 2026-2035
Si ~100 GPCRs siguen huérfanos a 2026, y cada deorphanization potencialmente abre una diana terapéutica nueva, el pipeline está lejos de agotarse. La inteligencia artificial aplicada a screening de ligandos (Chemistry42, Pharma.AI, AlphaFold-Multimer para predicción de complejos receptor-ligando) acelera drásticamente el proceso. La deorphanization de GFRAL en 2017 y de GPR75 en 2021 son ejemplos recientes con tracción clínica directa — ambos en obesidad, sector donde el éxito de los GLP-1 ha energizado la inversión industrial.
Preguntas frecuentes sobre GPCRs
¿Todos los receptores son GPCRs?
No. Los GPCRs son la familia más numerosa de receptores de membrana, pero hay al menos otras tres categorías funcionales mayores. Receptores ionotrópicos (canales iónicos ligando-dependientes): GABA-A, NMDA, AMPA, glicina, nicotínico de acetilcolina. Receptores tirosina kinasa (RTK): receptor de insulina, EGFR, VEGFR, factores de crecimiento. Receptores nucleares: receptor androgénico, estrogénico, tiroideo, glucocorticoide, vitamina D, PPAR. Cada familia opera con mecanismo distinto y está sujeta a farmacología propia. El error técnico más frecuente del lector divulgativo es asumir que cualquier «receptor» es GPCR — no lo es.
¿Cuál es la diferencia entre un GPCR y un receptor ionotrópico?
El receptor ionotrópico es un canal iónico que se abre directamente al unirse al ligando, modificando el potencial de membrana en milisegundos. El GPCR no es un canal: es una proteína que, al unirse al ligando, activa una cascada bioquímica intracelular cuyos efectos tardan segundos a minutos. La consecuencia farmacológica es radicalmente distinta: un agonista ionotrópico produce efecto inmediato (ketamina sobre NMDA en anestesia, benzodiacepina como PAM de GABA-A en sedación); un agonista GPCR produce efecto más lento pero más amplificado y sostenido.
¿Los GPCRs están solo en la membrana plasmática?
Mayoritariamente sí, pero no exclusivamente. Tras la activación y reclutamiento de β-arrestina, muchos GPCRs se internalizan en endosomas, donde pueden continuar señalizando vía β-arrestina con cinética más lenta y duradera. Además, se han descrito GPCRs activos en membranas intracelulares (núcleo, mitocondria), aunque el significado fisiológico está todavía en debate.
¿Cuántos fármacos actúan realmente sobre GPCRs?
Aproximadamente el 34% de los fármacos aprobados por la FDA tienen un GPCR como diana primaria (Hauser et al. Nat Rev Drug Discov 2017). Este porcentaje es estable en el tiempo. Otras dianas mayores: kinasas (~20%), receptores nucleares (~10%), canales iónicos (~5%), receptores enzimáticos no kinasa, transportadores, proteínas estructurales. El liderazgo de los GPCRs como diana farmacológica es estructural, no coyuntural.
¿Por qué la industria sigue invirtiendo tanto en GPCRs si ya hay tantos fármacos?
Cinco razones combinadas. Primera: ~100 GPCRs huérfanos sin diana caracterizada todavía. Segunda: el agonismo sesgado abre la posibilidad de mejorar fármacos existentes con mejor perfil seguridad-eficacia. Tercera: la modulación alostérica permite «tunear» receptores con techo de seguridad estructural. Cuarta: la dimerización y heterómeros descritos en los últimos años abren un espacio farmacológico completamente nuevo. Quinta: el éxito comercial monumental de los GLP-1 (~$50bn anuales combinados Eli Lilly + Novo Nordisk en 2024-2025) ha reorientado el flujo de I+D farmacéutico hacia receptores metabólicos — muchos GPCRs.
Sin entender GPCR, no se entiende la farmacología moderna de longevidad.
Este artículo es contenido editorial formativo. No sustituye al criterio médico individualizado. Las dosis, fármacos y ejemplos clínicos citados son referencias bibliográficas, no recomendaciones de prescripción. El estado regulatorio de los fármacos en pipeline mencionados puede haber cambiado tras la fecha de publicación; verifique siempre con la agencia regulatoria competente antes de cualquier decisión clínica.
- Fredriksson R, Lagerström MC, Lundin LG, Schiöth HB. The G-Protein-Coupled Receptors in the Human Genome Form Five Main Families. Phylogenetic Analysis, Paralogon Groups, and Fingerprints. Mol Pharmacol. 2003;63(6):1256-1272. PMID: 12761335. (Sistema de clasificación GRAFS).
- Bjarnadóttir TK, Gloriam DE, Hellstrand SH, et al. Comprehensive repertoire and phylogenetic analysis of the G protein-coupled receptors in human and mouse. Genomics. 2006;88(3):263-273. PMID: 16753280. (Cifras canónicas de GPCRs en genoma humano).
- Hauser AS, Attwood MM, Rask-Andersen M, Schiöth HB, Gloriam DE. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov. 2017;16(12):829-842. PMID: 29075003. (Porcentaje de fármacos FDA sobre GPCRs, pipeline).
- Rasmussen SG, DeVree BT, Zou Y, et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477(7366):549-555. PMID: 21772288. (Cristalografía del GPCR activado, base estructural del campo).
- Kenakin T. Biased Receptor Signaling in Drug Discovery. Pharmacol Rev. 2019;71(2):267-315. PMID: 30914442. (Marco teórico del agonismo sesgado).
- Mullican SE, Lin-Schmidt X, Chin CN, et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat Med. 2017;23(10):1150-1157. PMID: 28846098. (Deorphanization de GFRAL).
- Akbari P, Gilani A, Sosina O, et al. Sequencing of 640,000 exomes identifies GPR75 variants associated with protection from obesity. Science. 2021;373(6550):eabf8683. PMID: 34210852. (Identificación de GPR75 como diana antiobesidad).
- Liang YL, Khoshouei M, Radjainia M, et al. Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature. 2017;546(7656):118-123. PMID: 28437792. (Estructura del GLP-1R activado).
- Jastreboff AM, Aronne LJ, Ahmad NN, et al. Tirzepatide Once Weekly for the Treatment of Obesity (SURMOUNT-1). N Engl J Med. 2022;387(3):205-216. PMID: 35658024.
- Markham A. Oliceridine: First Approval. Drugs. 2020;80(16):1739-1744. PMID: 33049019. (Agonismo sesgado μ-opioide en clínica).
- Goodman & Gilman. The Pharmacological Basis of Therapeutics. 14th ed. McGraw-Hill; 2022. Capítulos 2-3 (Farmacología de receptores, GPCRs).
- Comité Nobel de Química. The Nobel Prize in Chemistry 2012. Otorgado a Robert J. Lefkowitz y Brian K. Kobilka por estudios sobre receptores acoplados a proteína G.