
Afinidad, potencia y eficacia: tres palabras que parecen sinónimos y no lo son
Confundirlas es el error farmacológico más caro de la práctica clínica. El fentanilo no es más eficaz que la morfina; es más potente. La diferencia ha matado a mucha gente.
Si solo te queda tiempo para tres conceptos de farmacología, son estos. Afinidad mide si el fármaco se une al receptor. Eficacia mide qué hace cuando se ha unido. Potencia mide cuánto fármaco hace falta. Tres ejes independientes. La industria entera, los Bios peptídicos y la mitad de los errores letales de prescripción se mueven sobre estos tres ejes.
Este es el cuarto artículo del cluster Glosario y cierra el bloque farmacológico fundamental que abren agonistas y antagonistas, vías de administración y receptores acoplados a proteína G. Si los tres primeros responden qué hace el fármaco, cómo llega y dónde actúa, este responde cuánto se necesita y qué efecto produce. Es la pieza más cuantitativa del cluster pero también la más rentable en clínica: la frontera entre prescribir bien y prescribir mal pasa casi siempre por entender bien estos tres parámetros.
El artículo cubre las tres definiciones formales (afinidad con Kd y Ki, eficacia intrínseca con Emax, potencia con EC50 e IC50), la curva dosis-respuesta como representación gráfica que los integra, la ventana terapéutica y el índice terapéutico como métricas de seguridad, los errores clínicos canónicos donde la confusión entre los tres tiene consecuencia letal, y la aplicación específica al pipeline de GLP-1 modernos donde el debate semaglutida-tirzepatida-retatrutida no se entiende sin esta gramática.
Por qué confundir estos tres conceptos es el error farmacológico más costoso
Afinidad, potencia y eficacia parecen sinónimos en lenguaje coloquial y no lo son en farmacología. La distinción es vieja, está perfectamente establecida en cualquier manual de Goodman & Gilman, y aún así es la fuente más recurrente de error en prescripción, en marketing farmacéutico y en debate público de medicamentos. Cuando alguien dice «el fentanilo es muy fuerte» no está distinguiendo si lo dice porque hace más efecto, porque hace falta menos cantidad para el mismo efecto, o porque se une más rápido al receptor. Son tres frases distintas que ocultan tres conceptos distintos.
La distinción que rara vez se explica bien
La forma más rápida de fijarla. Afinidad mide si un fármaco se une al receptor — es propiedad termodinámica del enlace ligando-receptor. Eficacia intrínseca mide qué hace ese enlace una vez establecido — es propiedad funcional del complejo ligando-receptor activado. Potencia mide cuánta cantidad de fármaco hace falta para obtener un efecto dado — es propiedad observable en la curva dosis-respuesta, que depende a la vez de la afinidad, de la eficacia, y de factores farmacocinéticos (absorción, distribución, metabolismo, eliminación).
Un fármaco puede tener afinidad altísima por su receptor (se une fuertemente) y eficacia cero — eso describe a cualquier antagonista neutro. Un fármaco puede tener afinidad modesta y eficacia máxima — puede ser un agonista completo que requiere concentraciones razonables para ocupar suficientes receptores. Un fármaco puede ser más potente que otro (basta menos cantidad para el mismo efecto) y tener idéntica eficacia (el efecto máximo alcanzable es el mismo).
El caso fentanilo-morfina como ejemplo paradigmático
El ejemplo más letal y mejor documentado de esta confusión. El fentanilo es entre 50 y 100 veces más potente que la morfina como analgésico — basta una dosis 50-100 veces menor en miligramos para producir analgesia equivalente. El carfentanilo, usado en veterinaria de grandes mamíferos, alcanza unas 10.000 veces la potencia de la morfina. Pero el techo de eficacia analgésica (la analgesia máxima alcanzable) es comparable entre los tres opioides — todos saturan el receptor mu-opioide. El fentanilo no es «más eficaz» que la morfina; es radicalmente más potente. La consecuencia clínica de no entender esta distinción es la base de la epidemia de sobredosis: dosificar fentanilo con la lógica de miligramos de la morfina mata directamente, porque la dosis euforizante de fentanilo está peligrosamente cerca de la dosis que produce depresión respiratoria, y el margen entre ambas es de un solo orden de magnitud.
Por qué importa al leer un Bio peptídico o un prospecto
Tres aplicaciones inmediatas para el lector KRECE. Primera: cuando un Bio o un fabricante reporta que un péptido nuevo es «más potente» que el anterior, eso no significa «más eficaz» ni «más seguro» — significa que basta menos cantidad para obtener un efecto. Puede ser bueno (mejor cumplimiento, menos volumen inyectado) o puede ser peor (ventana terapéutica más estrecha, mayor riesgo en errores de dilución). Segunda: cuando se compara la pérdida de peso de semaglutida vs tirzepatida vs retatrutida, no se compara «eficacia sobre el receptor GLP-1» sino «eficacia sobre el sistema metabólico integrado», que involucra varios receptores distintos. La sección 08 lo desarrolla. Tercera: cuando un prospecto indica una dosis máxima, esa cifra deriva de la curva dosis-respuesta del fármaco — pasarse no es «más eficaz», es más tóxico sin más beneficio.
Afinidad: la fuerza física de unión al receptor
La afinidad cuantifica la tendencia de un ligando a unirse a su receptor y permanecer unido. Es una propiedad puramente bioquímica, independiente de lo que el receptor haga después.
Definición técnica
La afinidad de un ligando por su receptor se define como la inversa de la constante de disociación del complejo ligando-receptor en equilibrio. Cuanto más estable es el enlace (mayor afinidad), menor concentración de ligando se necesita para mantener una fracción determinada de receptores ocupados. La unidad estándar es nanomolar (nM) o picomolar (pM) según el rango: ligandos con afinidad alta tienen Kd en orden de pM o sub-nM; ligandos con afinidad baja tienen Kd en orden de µM o mM.
La constante de disociación Kd
Es la métrica formal. Kd (constante de disociación) se define como la concentración de ligando a la cual el 50% de los receptores están ocupados en equilibrio. La fórmula deriva de la ley de acción de masas: si la concentración de ligando libre iguala a Kd, entonces la mitad de los sitios de unión están ocupados y la mitad libres. Cuanto menor es Kd, mayor es la afinidad. Un ligando con Kd = 1 nM se une 1000 veces más fuertemente que uno con Kd = 1 µM.
La constante de inhibición Ki
Es el análogo de Kd para ensayos de competición. En un experimento de binding competitivo, se mide cuánta concentración del ligando de interés se necesita para desplazar al 50% un ligando trazador de referencia preunido al receptor. La cifra resultante, una vez corregida por la concentración del trazador y su propia afinidad mediante la ecuación de Cheng-Prusoff, es la Ki. En la práctica clínica e investigadora, Kd y Ki suelen reportarse indistintamente como medidas de afinidad, aunque técnicamente Ki tiene la corrección del competidor incorporada.
Por qué afinidad alta no garantiza efecto biológico
Punto fundamental que se desliga del que más tarde explica el agonismo parcial e inverso. Un ligando puede tener una afinidad espectacular por su receptor y producir cero efecto biológico — es el caso de cualquier antagonista neutro. La afinidad solo mide la tenacidad del enlace; lo que hace el receptor cuando el ligando está unido es un fenómeno completamente independiente que llamamos eficacia intrínseca y desarrollamos en la siguiente sección. Afinidad y eficacia son variables independientes. Esta independencia es lo que hace posible que un único receptor tenga agonistas, agonistas parciales, antagonistas neutros y agonistas inversos, todos con afinidades similares pero con efectos cualitativamente distintos.
Antagonistas y agonistas pueden tener la misma Kd
Consecuencia directa de lo anterior. Tomemos el receptor mu-opioide. La morfina (agonista) y la naloxona (antagonista) tienen ambas alta afinidad por el receptor — sus Kd son del mismo orden. Lo que las separa cualitativamente es la eficacia intrínseca: la morfina activa el receptor (cascada de Gi/Go, disminución de cAMP, hiperpolarización neuronal, analgesia); la naloxona ocupa el sitio sin activar nada y, lo más relevante clínicamente, desplaza a la morfina o al fentanilo del receptor precisamente porque tiene afinidad equivalente o superior. La naloxona no funciona por ser «más fuerte»; funciona porque compite con afinidad equivalente sin producir efecto.
Eficacia intrínseca: la magnitud de la respuesta cuando el receptor está saturado
La eficacia mide qué hace el receptor cuando el ligando está unido. Es la propiedad funcional del complejo ligando-receptor que define el carácter cualitativo del fármaco: agonista, agonista parcial, antagonista neutro, agonista inverso.
Definición técnica de eficacia
La eficacia intrínseca (eficacia o actividad intrínseca, según notación) se define como la magnitud del efecto biológico que un complejo ligando-receptor produce por unidad de receptor activado. En términos operativos: la respuesta máxima alcanzable (Emax) cuando el ligando ocupa el 100% de los receptores disponibles, comparada con la respuesta máxima de un ligando de referencia (típicamente el agonista endógeno natural).
La escala convencional
Por convención, la eficacia se expresa como número entre -1 y +1 (o equivalentemente entre 0 y 1 para agonistas, con valores negativos para agonistas inversos):
| Tipo | Eficacia intrínseca | Comportamiento |
|---|---|---|
| Agonista completo | 1 (o 100%) | Reproduce la respuesta máxima del ligando endógeno |
| Super-agonista | > 1 | Respuesta superior a la del ligando endógeno (raro, ej. tirzepatida sobre GIPR) |
| Agonista parcial | Entre 0 y 1 | Respuesta sub-máxima incluso a saturación |
| Antagonista neutro | 0 | Ocupa el receptor sin generar respuesta |
| Agonista inverso | < 0 | Reduce la actividad basal espontánea del receptor |
Esta clasificación, recordada del cornerstone Agonistas y Antagonistas, depende enteramente del valor numérico de la eficacia intrínseca, no de la afinidad. Dos ligandos pueden tener idéntica Kd y caer en categorías distintas según su eficacia.
Por qué afinidad y eficacia son variables independientes
La consecuencia conceptual más importante de la sección anterior, reformulada desde el ángulo de la eficacia. La afinidad mide la tendencia del ligando a estar unido; la eficacia mide qué hace una vez unido. Operan en dos pasos secuenciales y separables del mecanismo molecular: primero acoplamiento (afinidad), después activación o no del receptor (eficacia). Un ligando puede ser tenaz en mantenerse unido (alta afinidad) y completamente inerte para activar la cascada de señalización (eficacia cero). Esto se diseña explícitamente en farmacología moderna — el agonismo sesgado (cubierto en el cornerstone Agonistas) consiste precisamente en mantener afinidad alta sobre el receptor mientras se modula selectivamente la eficacia hacia una u otra vía de señalización intracelular.
Reserva de receptor (spare receptors): la complicación que reordena la ecuación
El sistema biológico complica la relación lineal entre eficacia molecular y respuesta observable. En muchos tejidos, la respuesta máxima del órgano se alcanza cuando solo una fracción de los receptores está ocupada. El resto de receptores existen pero son «redundantes» para alcanzar el efecto máximo — son los llamados spare receptors o reserva de receptor.
La consecuencia cuantitativa es importante. Cuando hay reserva de receptor, la EC50 aparente del agonista (concentración que produce 50% de la respuesta del órgano) es menor que su Kd (concentración que ocupa el 50% de los receptores). El órgano alcanza su respuesta máxima con ocupación parcial de receptores; basta ocupar una fracción para que la cascada de amplificación intracelular llegue al techo del sistema. Este fenómeno es lo que explica que, en ciertas situaciones, dosis pequeñas de un agonista produzcan efectos clínicos plenos sin necesidad de saturar todos los receptores diana.
Ejemplo canónico: la respuesta cardíaca inotrópica a catecolaminas en corazón sano puede alcanzar Emax con apenas 10-20% de ocupación de receptores beta-1 — el 80-90% restante es reserva. Esto cambia drásticamente en insuficiencia cardíaca, donde la downregulation de receptores reduce o elimina la reserva y la curva dosis-respuesta se desplaza hacia la derecha (mayor EC50 aparente).
Potencia: la concentración necesaria para producir un efecto determinado
La potencia es la magnitud que más se confunde en lenguaje cotidiano con «eficacia» o «fuerza». Es estrictamente una medida de dosis: cuánta cantidad de fármaco hay que administrar para obtener un efecto dado.
Definición técnica de potencia
La potencia de un fármaco se mide como la concentración (o dosis) necesaria para producir un efecto de magnitud determinada — convencionalmente, el 50% del efecto máximo de ese fármaco. Cuanto menor es la concentración necesaria, mayor es la potencia. La potencia depende simultáneamente de la afinidad (capacidad de unirse), de la eficacia intrínseca (capacidad de activar el receptor), de la presencia o no de reserva de receptor, y, en clínica humana, de factores farmacocinéticos como biodisponibilidad, distribución y aclaramiento.
EC50: la concentración efectiva 50
La EC50 (effective concentration 50%) es la concentración del fármaco que produce el 50% de su propia respuesta máxima. Es la métrica gráfica que se lee en la curva dosis-respuesta: el punto del eje X correspondiente al cruce con la línea horizontal del 50% del Emax. Cuanto menor es la EC50 en mg/L, nM o µM, mayor es la potencia.
Punto sutil pero importante: la EC50 se refiere al 50% de la respuesta máxima del propio fármaco, no del sistema completo. Esto es lo que permite que un agonista parcial tenga una EC50 baja (alta potencia para alcanzar su propio techo sub-máximo) aunque su eficacia total sea inferior a la de un agonista completo.
IC50: la concentración inhibitoria 50
Análogo de la EC50 para antagonistas e inhibidores. La IC50 (inhibitory concentration 50%) es la concentración del antagonista o inhibidor que reduce al 50% una respuesta biológica medida (frente a la respuesta basal o frente a la respuesta inducida por un agonista de referencia). Se usa universalmente en ensayos farmacológicos in vitro para caracterizar antagonistas, inhibidores enzimáticos y bloqueadores. En la curva dosis-respuesta del antagonista, la IC50 marca el punto donde la inhibición es media.
Por qué potencia NO es eficacia
La distinción más cara de la farmacología clínica. Repetida desde la sección 01 pero ahora cuantificable: dos fármacos pueden tener Emax idéntico (misma eficacia máxima alcanzable) y EC50 muy distintos (muy distinta potencia). En la curva dosis-respuesta, esto se ve como dos curvas con la misma meseta pero desplazadas horizontalmente. El más potente alcanza su Emax a concentración menor; el menos potente lo alcanza también, pero a concentración mayor.
Inversamente, dos fármacos pueden tener idéntica EC50 (misma potencia) y Emax distintos (distinta eficacia). En la curva, son dos curvas con el mismo desplazamiento horizontal pero distinta altura de meseta. La sección 05 desarrolla la lectura gráfica completa.
Equipotencia: cómo se construyen las tablas de conversión clínica
Cuando dos fármacos actúan sobre el mismo receptor con eficacia comparable pero potencia distinta, se pueden establecer dosis equipotentes: la cantidad de cada uno que produce un efecto clínico equivalente. Estas tablas son herramienta operativa fundamental para switch de fármacos en práctica clínica. Tres ejemplos canónicos:
Opioides: 10 mg de morfina intravenosa equivalen aproximadamente a 100 µg de fentanilo IV, 1 mg de hidromorfona IV, y unos 100 mg de tramadol oral. Las tablas de conversión son herramienta diaria en oncología, paliativos y dolor crónico.
Benzodiacepinas: aproximadamente 10 mg de diazepam equivalen a 1 mg de alprazolam, 1-2 mg de lorazepam, 0.5 mg de clonazepam. Estas equivalencias se usan rutinariamente en deshabituación, switching y desescalada — error en la conversión es causa frecuente de síndrome de abstinencia o sobredosificación.
Corticoides: aproximadamente 5 mg de prednisona equivalen a 4 mg de metilprednisolona, 0.75 mg de dexametasona, 20 mg de hidrocortisona. Esencial en pulsos, supresión adrenal y manejo perioperatorio.
La construcción de tablas de equipotencia requiere ensayos clínicos comparativos head-to-head; cuando no existen, se infieren de datos farmacodinámicos y se usan con cautela.
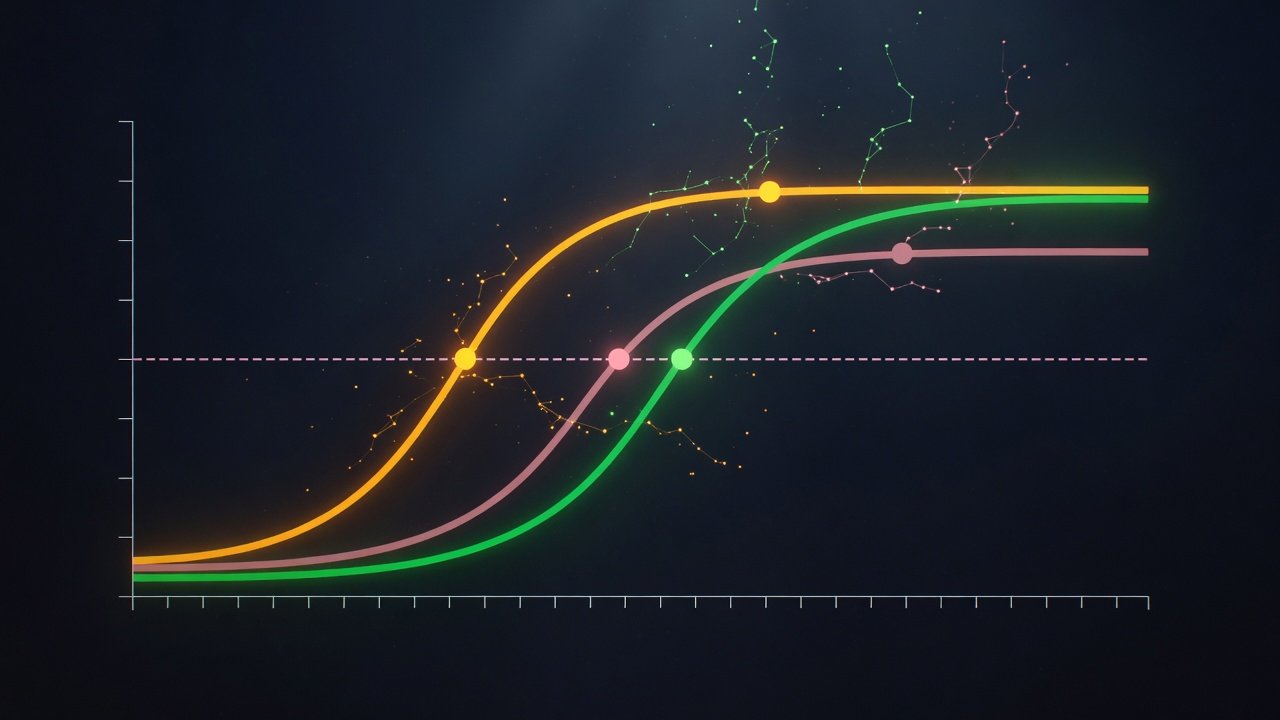
La curva dosis-respuesta: cómo se grafica todo lo anterior
Toda la conversación cuantitativa de afinidad, potencia y eficacia se condensa en un gráfico estándar que todo prescriptor debería saber leer.
Forma sigmoidea y por qué el eje X es logarítmico
La curva dosis-respuesta canónica representa en el eje Y la magnitud de la respuesta (en valor absoluto o como porcentaje del Emax), y en el eje X la concentración o dosis del fármaco, expresada en escala logarítmica. La forma resultante es una S sigmoidea característica: una fase inicial plana (dosis por debajo del umbral, ningún efecto observable), una fase lineal de ascenso (relación aproximadamente lineal entre log de concentración y respuesta), y una meseta (Emax, donde aumentar la dosis ya no aumenta el efecto).
La razón del eje X logarítmico es práctica: los efectos farmacológicos se distribuyen en un rango amplísimo de concentración (típicamente 3-4 órdenes de magnitud entre el umbral y la saturación), y representarlos en escala lineal comprimiría visualmente toda la fase activa contra el origen. El log convierte la sigmoide en una curva legible donde se distingue claramente cada parámetro.
Cómo se lee EC50 en la curva
EC50 es el punto del eje X donde la curva cruza el 50% del Emax. Operacionalmente: se localiza la altura de meseta (Emax), se baja a la mitad en el eje Y, se traza una línea horizontal hasta cortar la curva, y desde ese punto se baja al eje X — la coordenada resultante es la EC50. Más a la izquierda en el eje X = menor EC50 = mayor potencia.
Comparar dos fármacos: curvas paralelas, desplazadas y divergentes
Cuatro patrones gráficos que el lector debe reconocer al comparar fármacos:
Curvas paralelas desplazadas horizontalmente — misma Emax, distinta EC50. Significa misma eficacia, distinta potencia. Ejemplo: morfina y fentanilo como analgésicos en condiciones equipotentes. La curva del fentanilo está desplazada a la izquierda (menos concentración para mismo efecto), pero ambas alcanzan la misma analgesia máxima.
Curvas con misma EC50 pero distinta altura de meseta — misma potencia, distinta eficacia. Ejemplo: agonista completo vs agonista parcial sobre el mismo receptor. Ambas curvas suben en el mismo punto del eje X, pero la del parcial se aplana antes.
Curvas divergentes — distinta EC50 y distinta Emax. Comparación entre fármacos con afinidades y eficacias distintas. La mayoría de comparaciones reales caen aquí.
Curvas desplazadas y aplanadas — el caso del antagonismo, que tiene patrones específicos:
Cómo se distingue gráficamente un antagonista competitivo de uno no competitivo
Este es el aprendizaje gráfico más útil del bloque farmacológico. Concepto recordado del cornerstone GPCR y ahora visible en la curva:
Antagonista competitivo reversible: al añadir antagonista a un sistema con agonista, la curva del agonista se desplaza hacia la derecha de forma paralela — mantiene la misma Emax (la respuesta máxima sigue siendo alcanzable si se aumenta suficientemente la concentración del agonista) y mantiene la pendiente (la cinética del sistema no cambia), solo aumenta la EC50 aparente. Cuanto más antagonista, más desplazamiento. Es el patrón clásico de naloxona sobre la curva analgésica de morfina, o de propranolol sobre la curva inotrópica de adrenalina.
Antagonista no competitivo o irreversible: al añadir antagonista, la curva del agonista baja su meseta — el Emax se reduce porque hay menos receptores funcionales disponibles, independientemente de cuánto agonista se administre. La EC50 puede permanecer aproximadamente igual (los receptores remanentes responden con su sensibilidad original), pero el techo del sistema cae. Patrón clásico de antagonistas alostéricos negativos y de bloqueadores covalentes como aspirina sobre COX-1 en plaquetas.
Una sola mirada a una curva dosis-respuesta con antagonista permite caracterizar inmediatamente el tipo de antagonismo: desplazamiento paralelo a la derecha = competitivo; meseta más baja = no competitivo o irreversible.
Curva de Schild como herramienta para caracterizar antagonismo
El método cuantitativo formal para caracterizar antagonismo competitivo. Se desarrolla H. O. Schild en los años 40 y permanece como referencia.
Operativamente: se mide la EC50 del agonista en ausencia de antagonista y en presencia de varias concentraciones crecientes de antagonista. Se calcula la dose ratio (DR): cociente entre la EC50 con antagonista y la EC50 sin antagonista. Se grafica el log(DR-1) en el eje Y frente al log de la concentración de antagonista en el eje X. Para un antagonismo competitivo puro, el resultado es una línea recta de pendiente 1. La intersección con el eje X define el parámetro pA2, que es el log negativo de la concentración de antagonista que dobla la EC50 (DR=2). El pA2 estima la afinidad del antagonista por el receptor: cuanto mayor pA2, mayor afinidad.
Este análisis es herramienta de investigación farmacológica más que de clínica práctica, pero define el lenguaje cuantitativo del campo. Cuando una publicación caracteriza un nuevo antagonista, el pA2 y la pendiente de Schild son métricas estándar; pendientes significativamente distintas de 1 indican que el antagonismo no es puramente competitivo.
Ventana terapéutica e índice terapéutico
Los conceptos anteriores describen cuánto efecto produce un fármaco a una dosis dada. La ventana terapéutica describe cuánto margen hay entre el efecto deseado y la toxicidad. Es la métrica de seguridad que ordena la prescripción real.
Definición de ventana terapéutica
La ventana terapéutica es el rango de concentraciones plasmáticas (o dosis) entre la dosis mínima eficaz (umbral inferior) y la dosis mínima tóxica (umbral superior). Es el espacio operativo donde el fármaco produce el efecto deseado sin desencadenar toxicidad relevante. Cuanto más ancha la ventana, más permisiva la dosificación y menor el riesgo de iatrogenia por errores.
LD50, ED50 y el índice terapéutico
El cociente cuantitativo entre toxicidad y eficacia se llama índice terapéutico (TI):
TI = LD50 / ED50
Donde LD50 es la dosis letal para el 50% de la población (medible en animales; en humanos se aproxima mediante TD50, dosis tóxica) y ED50 es la dosis eficaz para el 50%. Un TI alto significa que la dosis tóxica está muy por encima de la eficaz — fármaco seguro. Un TI bajo significa que la dosis tóxica está apenas por encima de la eficaz — fármaco peligroso, requiere monitorización estricta.
Tabla de índice terapéutico de fármacos canónicos
| Fármaco | TI aproximado | Implicación clínica |
|---|---|---|
| Digoxina | ~2 | Ventana extremadamente estrecha. Beneficio inotrópico y arritmia letal separadas por mínimo margen |
| Litio | ~2-3 | Variaciones de excreción renal disparan toxicidad neurológica |
| Fentanilo | ~2 | Dosis euforizante y dosis de depresión respiratoria casi idénticas |
| Warfarina | ~2-5 | Control de INR obligatorio; cambios de dieta o interacciones disparan hemorragia o trombosis |
| Paracetamol | ~10-20 | Dosis estándar 1 g; LD50 hepática 10-20 g. Suficientemente accesible para autolisis |
| Benzodiacepinas | > 100 | Difícil mortalidad por sobredosis aislada; razón por la que reemplazaron a barbitúricos |
| Penicilina | > 100 | Riesgo principal es anafilaxia, no toxicidad por dosis |
Por qué importa el margen, no la dosis absoluta
El TI clarifica una intuición clínica importante: la peligrosidad de un fármaco no se mide por la dosis prescrita sino por el cociente entre dosis tóxica y dosis útil. Un fármaco potente con TI alto es más seguro que un fármaco débil con TI bajo. El fentanilo y la digoxina ejemplifican lo segundo: dosis pequeñas pero con margen letal mínimo. Las benzodiacepinas ejemplifican lo primero: dosis modestas en mg con margen amplísimo entre eficacia y toxicidad letal en monoterapia.
Implicación operativa: los fármacos con TI bajo requieren monitorización terapéutica activa (niveles plasmáticos en digoxina, litio; INR en warfarina) porque la dosis óptima no se puede inferir de la dosis administrada sola — depende de absorción, distribución, metabolismo y eliminación individuales del paciente. Este es uno de los puntos donde la práctica clínica deja de ser «prescribir miligramos» y pasa a ser «ajustar concentración plasmática individual».
Errores clínicos canónicos por confusión de parámetros
Cuatro casos documentados donde la confusión entre afinidad, potencia y eficacia ha causado morbimortalidad significativa y persistente. Cada uno enseña una lección distinta.
Fentanilo vs morfina: cuando la potencia mata
El caso paradigmático ya esbozado. El fentanilo es 50-100 veces más potente que la morfina como analgésico. La dosis equivalente para analgesia es del orden de microgramos en lugar de miligramos. Pero el techo de eficacia analgésica es similar: ambos saturan el receptor mu-opioide. El error clínico recurrente es interpretar la mayor potencia como mayor «fuerza» y dosificar con la lógica métrica de la morfina. La consecuencia: dosis euforizantes y dosis letales por depresión respiratoria separadas por un factor de 2-3 en lugar de los 10-20 habituales en morfina. El fentanilo no es más eficaz; es más potente, y eso lo hace más peligroso por unidad de masa. El carfentanilo, 10.000 veces más potente que la morfina, lleva esta lógica al extremo y es la base de muchas muertes accidentales en consumidores que esperaban heroína.
Calcifediol vs colecalciferol: cuando las unidades engañan
Caso recurrente en consulta clínica española dada la disponibilidad de calcifediol como Hidroferol® (Faes Farma) en farmacia. El colecalciferol (vitamina D3, forma estándar de suplementación oral) y el calcifediol (25-hidroxivitamina D3, forma activa intermedia) no son intercambiables en dosis equivalente: el calcifediol es aproximadamente 3 a 5 veces más potente porque salta el paso de hidroxilación hepática y eleva más rápidamente los niveles séricos de 25-OH-D.
El error documentado: prescriptor confunde unidades y dosifica calcifediol con las cifras habituales de colecalciferol (1.000-5.000 UI/día o equivalente), produciendo elevación suprafisiológica de 25-OH-D, hipercalcemia clínica e insuficiencia renal aguda en casos extremos. La práctica clínica recomienda mantener el colecalciferol como suplemento de elección para la mayoría de pacientes y reservar el calcifediol para situaciones específicas (malabsorción, insuficiencia hepática) con dosificación y monitorización estrechas. Sobre los umbrales óptimos de 25-OH-D y las dosis recomendadas hay debate activo entre paneles internacionales (IOM/NAM 2010, Endocrine Society, EFSA), y este artículo no pretende resolverlo — solo recordar que calcifediol y colecalciferol no se dosifican con la misma cifra.
DHT vs testosterona: afinidad alta sin eficacia uniforme entre tejidos
Caso pedagógicamente interesante. La dihidrotestosterona (DHT) tiene afinidad por el receptor androgénico aproximadamente 5 veces superior a la testosterona, y eficacia transcripcional también superior en tejidos donde está presente (próstata, folículo piloso, piel). Pero la DHT solo se produce localmente por la 5-alfa-reductasa en esos tejidos específicos, no circula en cantidades comparables a la testosterona como hormona sistémica.
La consecuencia clínica: la finasterida y dutasterida, inhibidores de la 5-alfa-reductasa, reducen drásticamente la DHT (hasta 70-90%) mientras elevan la testosterona total un 10-30%. Resultado tisular heterogéneo: efecto antiandrogénico marcado en próstata y folículo piloso (de ahí la indicación en HBP y alopecia androgénica), pero efectos androgénicos sistémicos preservados o aumentados en músculo y otros tejidos. El mismo eje hormonal con dos ligandos de afinidad y eficacia muy distintas en tejidos distintos — ejemplo limpio de cómo un mismo receptor responde de forma heterogénea según contexto local.
AINEs a dosis máxima: subir dosis no sube eficacia, sube toxicidad
El error de prescripción más común en farmacia comunitaria y consulta primaria. El ibuprofeno tiene un techo de eficacia analgésica en torno a los 400-600 mg por dosis; subir a 800 o 1.200 mg no produce analgesia adicional clínicamente relevante pero sí incrementa proporcionalmente el riesgo gastrointestinal, renal y cardiovascular. La curva dosis-respuesta del ibuprofeno para analgesia tiene meseta antes de los 600 mg; la curva dosis-respuesta para toxicidad es lineal hasta dosis mucho mayores.
Implicación clínica: la práctica de «subir la dosis hasta que funcione» en dolor agudo con AINEs es farmacológicamente errónea — si 600 mg no analgesian, 1.200 mg tampoco lo harán mejor pero sí duplicarán riesgo gástrico y renal. La combinación adicional con IECA/ARA-II y diuréticos —el llamado «Triple Whammy» en literatura nefrológica— multiplica por aproximadamente 10 el riesgo de insuficiencia renal aguda, especialmente en mayores y pacientes con función renal limítrofe.
Benzodiacepinas equipotentes: tabla de conversión y rebotes
Quinto caso, especialmente relevante en geriatría y psiquiatría. Las benzodiacepinas comparten eficacia (todas son moduladores alostéricos positivos del receptor GABA-A) pero difieren enormemente en potencia (mg requerido para efecto equivalente) y en vida media (T½ desde 6 horas para alprazolam hasta más de 100 horas para diazepam y sus metabolitos activos).
El error documentado: switch de una benzodiacepina a otra sin aplicar la equivalencia de potencia, produciendo infradosis (síndrome de abstinencia con ansiedad rebote, insomnio, convulsiones en casos severos) o sobredosis (sedación profunda, caídas, deterioro cognitivo agudo en ancianos). La regla práctica clínica para deshabituación incluye el switching de benzodiacepinas de vida media corta y alta potencia (alprazolam) a diazepam para permitir desescalada lenta del 10-25% de dosis cada 2-4 semanas, según protocolo. Las tablas de equivalencia son herramienta operativa cotidiana en deshabituación benzodiacepínica.
Por qué estos parámetros importan en longevidad y péptidos
El debate semaglutida vs tirzepatida vs retatrutida es el caso de uso más relevante del cluster para el lector KRECE, y solo se entiende con la gramática de las secciones anteriores.
GLP-1: semaglutida vs tirzepatida vs retatrutida — más receptores, no más eficacia
La comparación clínica de magnitud de pérdida de peso es conocida: semaglutida aproximadamente 15% (STEP-1), tirzepatida hasta 22.5% (SURMOUNT-1 a 72 semanas con 15 mg), retatrutida aproximadamente 24-25% en ensayos fase 2. La interpretación intuitiva sería «tirzepatida y retatrutida son más eficaces que semaglutida». Es una verdad observable pero parcialmente engañosa cuando se examina a nivel de receptor.
Sobre el receptor GLP-1R aislado, la semaglutida tiene mayor potencia molecular que la tirzepatida. Los datos publicados de Coskun et al. (2018) en el desarrollo de tirzepatida son inequívocos: la semaglutida tiene aproximadamente la mitad de la potencia del GLP-1 nativo sobre GLP-1R, mientras que la tirzepatida tiene aproximadamente un cuarto de la potencia del GLP-1 nativo sobre el mismo receptor — la mitad que la semaglutida. Sobre el receptor GIP (GIPR), en cambio, la tirzepatida tiene potencia comparable a la del GIP nativo (~1×), mientras que la semaglutida es esencialmente inerte. Es decir: tirzepatida no supera a semaglutida en GLP-1R — la subactiva deliberadamente — y añade un segundo receptor que la semaglutida no toca.
La ganancia clínica de tirzepatida sobre semaglutida no es por «más eficaz sobre GLP-1R» sino por activación adicional del GIPR con potencia equivalente a la hormona nativa, que aporta efectos metabólicos complementarios (acción sobre adipocito, mejor tolerancia gastrointestinal porque el GIP modula la respuesta de náusea en área postrema) y permite mayor escalada de dosis sin saturar efectos adversos. Retatrutida añade además agonismo sobre el receptor de glucagón (GCGR), que aumenta el gasto energético basal por aceleración del metabolismo hepático lipídico — y su componente sobre GLP-1R es menos potente todavía que el de tirzepatida, lo que confirma que el diseño no busca máxima activación de GLP-1R sino activación equilibrada de tres receptores complementarios.
La lección farmacológica es clara: se está optimizando el efecto clínico sumando receptores, no maximizando la potencia sobre un solo receptor. Esta es la lógica detrás de toda la siguiente generación de polidagonistas en pipeline (cagrisema en GLP-1 + amilina, agonistas GLP-1 + glucagón sin GIP, etc.).
Por qué un super-agonista puede tener menor potencia que un agonista completo
Caso técnico que confunde a muchos lectores. Un super-agonista es un ligando que produce respuesta superior al ligando endógeno natural — eficacia intrínseca > 1 (sección 03). Pero esa caracterización es sobre eficacia, no sobre potencia. Un super-agonista puede requerir concentraciones mayores que el ligando endógeno para alcanzar su pico (EC50 más alta = menor potencia) y aún así producir un efecto máximo superior. Tirzepatida sobre GIPR ilustra el extremo: eficacia ligeramente superior al GIP nativo, pero potencia (EC50) en rango similar. No es «más fuerte»; es «alcanza un techo distinto».
GHSR y la diferencia entre ghrelina nativa y secretagogos farmacológicos
El receptor GHSR (growth hormone secretagogue receptor, receptor de ghrelina, GPCR clase A) ilustra otro caso de disociación afinidad-eficacia-potencia. Su ligando endógeno es la ghrelina (péptido de 28 aminoácidos acilado). Los secretagogos farmacológicos incluyen péptidos sintéticos (ipamorelina, GHRP-6, hexarelina) y un secretagogo oral no peptídico, MK-677 (ibutamoren).
La ipamorelina tiene afinidad y eficacia comparables a la ghrelina sobre GHSR, pero su selectividad por la vía Gq sin reclutamiento significativo de beta-arrestina la hace producir menos desensibilización con uso continuado (ejemplo de agonismo sesgado aplicado, recordado del cornerstone Agonistas). MK-677 tiene afinidad menor pero biodisponibilidad oral, lo que cambia el perfil farmacocinético sin necesariamente cambiar el perfil farmacodinámico. La elección clínica entre estos secretagogos no se hace por «cuál es más potente», sino por el balance entre selectividad de vía, tolerancia crónica y vía de administración.
Modulación alostérica: PAM/NAM cambian EC50 sin tocar Emax
Concepto recordado del cornerstone Agonistas y Antagonistas y ahora aplicable cuantitativamente. Un modulador alostérico positivo (PAM) se une a un sitio distinto del ortostérico y aumenta la afinidad y/o la eficacia del ligando endógeno por su receptor. El efecto neto sobre la curva dosis-respuesta es: baja la EC50 aparente del ligando endógeno sin modificar significativamente su Emax. En lenguaje gráfico: desplaza la curva hacia la izquierda, manteniendo la meseta.
Las benzodiacepinas son el ejemplo paradigmático: como PAM del receptor GABA-A, no abren el canal de cloro por sí solas (no tienen eficacia agonista directa), pero potencian masivamente el efecto del GABA endógeno cuando éste se libera. La consecuencia farmacológica es un techo de seguridad estructural — no es posible «sobreestimular» el sistema GABAérgico con benzodiacepinas en monoterapia porque la respuesta sigue dependiendo del GABA endógeno disponible. Esta es la razón mecanística del TI >100 de las benzos.
El cinacalcet (Sensipar®/Mimpara®, aprobado FDA 2004) es PAM del receptor sensor de calcio (CaSR, GPCR clase C) en paratiroides: sensibiliza el receptor al calcio extracelular, induciendo supresión de PTH a niveles de calcio que normalmente no la suprimirían. Uso clínico en hiperparatiroidismo secundario en pacientes en hemodiálisis y en carcinoma paratiroideo.
Los moduladores alostéricos negativos (NAM) hacen lo opuesto: desplazan la curva del agonista hacia la derecha (suben EC50 aparente) sin modificar la Emax intrínseca del sistema. Ejemplos en pipeline psiquiátrico (mGluR5 NAM en X frágil, mGluR2/3 NAM en depresión resistente) sin aprobaciones clínicamente relevantes a 2026.
Microdosing: jugar con la curva, no con la dosis absoluta
Caso aplicado al lector KRECE. La práctica del microdosing de péptidos — usar dosis significativamente menores que las del rango terapéutico estándar — se justifica farmacológicamente cuando hay reserva de receptor suficiente para alcanzar efecto clínico con ocupación parcial, o cuando se busca evitar la desensibilización por uso crónico de dosis máximas. Aplicado a GLP-1, microdosing es uso por debajo de dosis máxima (típicamente 0.25-0.5 mg semanal en lugar de los 1-2.4 mg de Wegovy) buscando facilitar transición de hábitos dietéticos sin saturación de receptores y sin efecto rebote masivo al discontinuar.
La justificación farmacológica del microdosing no es «menos es mejor» sino «menos puede ser suficiente cuando hay reserva de receptor y se trabaja en la fase ascendente de la curva dosis-respuesta, antes de la meseta». Es una decisión que requiere entender la curva del fármaco concreto y monitorizar la respuesta individual — no una recomendación general aplicable a cualquier péptido o suplemento.
Preguntas frecuentes
¿Más potente significa más eficaz?
No. Potencia y eficacia son ejes independientes. Más potente significa que basta menos cantidad para producir un efecto; más eficaz significa que el efecto máximo alcanzable es mayor. Un fármaco puede ser más potente y menos eficaz, o más potente y similarmente eficaz. El fentanilo es radicalmente más potente que la morfina, pero su techo de eficacia analgésica es comparable.
¿Por qué dos fármacos con la misma Kd pueden hacer cosas opuestas?
Porque afinidad (Kd) y eficacia intrínseca son variables independientes. La afinidad mide solo si el fármaco se une al receptor y con qué tenacidad; la eficacia mide qué hace el receptor cuando está unido. Un agonista y un antagonista neutro pueden tener Kd idéntica — se unen ambos con la misma fuerza — pero solo el agonista activa la cascada intracelular. El antagonista ocupa el sitio y bloquea, sin producir efecto.
¿Es mejor dosis baja de fármaco muy potente o dosis alta de fármaco menos potente?
Depende. La pregunta correcta no es sobre la dosis sino sobre el índice terapéutico de cada fármaco. Un fármaco muy potente con índice terapéutico bajo (fentanilo, digoxina) es más peligroso por unidad de masa que uno menos potente con índice terapéutico alto (penicilina, benzodiacepinas). La elección clínica entre dos fármacos para la misma indicación se hace sobre potencia, eficacia, perfil de efectos secundarios, vida media, vía de administración disponible y ventana terapéutica — no solo sobre potencia.
¿Cómo afecta esto a la decisión de cambiar de un GLP-1 a otro?
El switch entre semaglutida, tirzepatida y otros agonistas GLP-1 no se hace por equipotencia en mg (no hay equivalencia simple) sino por escalada progresiva titulando según tolerancia y respuesta clínica individual. La tirzepatida tiene mayor magnitud de pérdida de peso clínica que la semaglutida no por mayor potencia GLP-1R sino por agonismo dual GLP-1R/GIPR. El cambio entre ambas requiere reiniciar titulación porque el receptor adicional (GIPR) modifica el perfil de efectos secundarios y de respuesta, no por intercambiabilidad farmacológica simple.
¿Cuándo conviene un agonista parcial frente a un agonista completo?
Tres situaciones clínicas paradigmáticas. Primera: cuando se necesita un techo de eficacia más bajo por seguridad — buprenorfina como agonista parcial mu-opioide para deshabituación de opioides en adicción, con menor riesgo de depresión respiratoria que metadona o morfina. Segunda: cuando se busca antagonismo funcional sin abolición total — un agonista parcial en presencia de un agonista endógeno hiperactivo se comporta como antagonista relativo. Tercera: cuando hay variabilidad de tono basal del receptor entre pacientes — el agonista parcial produce respuesta más uniforme que el agonista completo en sistemas con eficacia constitutiva variable. La pimavanserina (agonista inverso 5-HT2A para psicosis Parkinson) y la buprenorfina son los ejemplos clínicos mejor documentados.
Sin manejar afinidad, potencia y eficacia, todo Bio peptídico se lee como folklore.
Este artículo es contenido editorial formativo. No sustituye al criterio médico individualizado. Las dosis, fármacos y ejemplos clínicos citados son referencias bibliográficas, no recomendaciones de prescripción. La conversión entre fármacos equipotentes (opioides, benzodiacepinas, corticoides) requiere supervisión médica directa por el riesgo de error en switching. La discusión sobre vitamina D, calcifediol y umbrales óptimos sigue siendo objeto de debate científico activo entre paneles internacionales y este artículo no pretende resolverlo.
- Goodman & Gilman. The Pharmacological Basis of Therapeutics. 14th ed. McGraw-Hill; 2022. Capítulo 3 (Farmacodinamia: mecanismos moleculares de acción de los fármacos).
- Kenakin T. A pharmacology primer: techniques for more effective and strategic drug discovery. 5th ed. Academic Press; 2019. Capítulos 2-5 (afinidad, potencia, eficacia, modelos cuantitativos).
- Coskun T, Sloop KW, Loghin C, et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: From discovery to clinical proof of concept. Mol Metab. 2018;18:3-14. PMID: 30473097. (Caracterización de tirzepatida sobre GLP-1R vs GIPR).
- Jastreboff AM, Aronne LJ, Ahmad NN, et al. Tirzepatide Once Weekly for the Treatment of Obesity (SURMOUNT-1). N Engl J Med. 2022;387(3):205-216. PMID: 35658024.
- Jastreboff AM, Kaplan LM, Frias JP, et al. Triple-Hormone-Receptor Agonist Retatrutide for Obesity — A Phase 2 Trial. N Engl J Med. 2023;389(6):514-526. PMID: 37366315.
- Wilding JPH, Batterham RL, Calanna S, et al. Once-Weekly Semaglutide in Adults with Overweight or Obesity (STEP-1). N Engl J Med. 2021;384(11):989-1002. PMID: 33567185.
- Schild HO. pA, a new scale for the measurement of drug antagonism. Br J Pharmacol Chemother. 1947;2(3):189-206. PMID: 20258355. (Análisis de Schild original).
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22(23):3099-3108. PMID: 4202581. (Ecuación de Cheng-Prusoff).
- Lindberg JS, Culleton B, Wong G, et al. Cinacalcet HCl, an oral calcimimetic agent for the treatment of secondary hyperparathyroidism in hemodialysis and peritoneal dialysis: a randomized, double-blind, multicenter study. J Am Soc Nephrol. 2005;16(3):800-807. PMID: 15689407. (Cinacalcet como PAM del CaSR).
- Moore N, Pollack C, Butkerait P. Adverse drug reactions and drug-drug interactions with over-the-counter NSAIDs. Ther Clin Risk Manag. 2015;11:1061-1075. PMID: 26203254. (AINEs, techo de eficacia analgésica, Triple Whammy renal).
- Aspirin in Heart Failure (AHF) Investigators, et al. EFSA Panel on Dietetic Products, Nutrition and Allergies. Scientific Opinion on the Tolerable Upper Intake Level of vitamin D. EFSA Journal. 2012;10(7):2813. (TUL EFSA 4.000 UI/día).
- Holick MF, Binkley NC, Bischoff-Ferrari HA, et al. Evaluation, Treatment, and Prevention of Vitamin D Deficiency: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011;96(7):1911-1930. PMID: 21646368. (Recomendaciones Endocrine Society, distinción colecalciferol vs calcifediol).