Calidad de los péptidos: qué es, cómo se mide y qué exigir antes de comprar
Cuatro dimensiones simultáneas — pureza, identidad, esterilidad y endotoxinas — que ningún método aislado puede cubrir. El estándar que acepta el comprador define el mercado.
La calidad de un péptido no es un número. Es una matriz de cuatro dimensiones — pureza, identidad, esterilidad y endotoxinas — que ningún método analítico aislado puede cubrir. Cualquier proveedor que reduzca esa matriz a un único valor (típicamente «99% HPLC-UV») está transfiriendo el riesgo al comprador. En 2025, según datos de testeo independiente del mercado, el 74% de los viales GLP-1 estaba fuera de especificación de potencia, el 12,7% de las muestras falló esterilidad y el 18% superó el límite de cuantificación de endotoxinas. El estándar que acepta el comprador define el mercado.
Este artículo es el pillar conceptual del cluster de calidad peptídica de KRECE. No es un caso de estudio — ya hay un caso de estudio sobre SS-31 — ni una pieza técnica sobre cómo leer un certificado de análisis — ya existe esa pieza técnica. Es el mapa que ordena el territorio: qué significa exactamente la palabra calidad cuando se aplica a un péptido terapéutico, qué métodos analíticos responden cada pregunta, qué datos cuantitativos describen el estado real del mercado en 2026 y qué criterio operativo aplica KRECE antes de aceptar un lote.
El artículo asume conocimiento básico de qué es un péptido terapéutico. Si esa base falta, el pillar editorial pertinente está en la taxonomía KRECE de péptidos. Tampoco intenta agotar la química analítica de proteinoides — esa es una disciplina entera. Lo que sí hace es delimitar las cuatro dimensiones simultáneas que componen la calidad de un péptido, traducir cada una a método analítico concreto, y publicar los datos de mercado que el comprador necesita para decidir.
¿Qué es la calidad de un péptido?
Definición operativa: la matriz de cuatro dimensiones
La calidad de un péptido es la combinación simultánea, verificable por lote, de cuatro propiedades independientes: (1) que el vial contenga mayoritariamente la molécula declarada y no productos de degradación o subproductos de síntesis — pureza; (2) que esa molécula mayoritaria sea exactamente el péptido nombrado en la etiqueta y no un isómero o un análogo de masa similar — identidad; (3) que el contenido del vial esté libre de microorganismos viables — esterilidad; y (4) que no contenga niveles clínicamente relevantes de pirógenos bacterianos — endotoxinas. Las cuatro son condiciones independientes. Cada una se verifica con un método analítico distinto y cada una puede fallar mientras las otras tres pasan.
Esta es la definición operativa que KRECE aplica al filtrar proveedores. No es una posición editorial original: es el estándar farmacéutico vigente, codificado en las farmacopeas USP (Estados Unidos) y Ph. Eur. (Europa), aplicado a cualquier producto biológico inyectable. La diferencia está en cómo se aplica fuera del ámbito farmacéutico regulado — donde la mayor parte del mercado peptídico opera hoy.
Pureza, identidad, potencia y seguridad biológica: cuatro cosas distintas
El error de marketing más frecuente es usar pureza como sinónimo de calidad. No lo es. Un péptido puede tener 99% de pureza cromatográfica y al mismo tiempo no contener apenas péptido activo si el peso del vial corresponde mayoritariamente a contraiones y agua residual. Puede pasar la pureza y fallar la identidad si lo que el cromatógrafo midió es un análogo de masa similar. Puede pasar pureza e identidad y fallar esterilidad si el llenado aséptico fue deficiente. Y puede pasar las tres anteriores y contener endotoxinas a niveles pirogénicos sin que ningún cromatograma lo detecte. Cada propiedad responde a una pregunta distinta y exige su método propio.
¿Qué significa «peptide quality» en la industria farmacéutica?
El término peptide quality en el léxico farmacéutico anglosajón cubre el mismo concepto matricial. La diferencia es contextual: en un entorno cGMP (current Good Manufacturing Practice) la calidad se da por descontada porque las cuatro dimensiones se verifican obligatoriamente por lote bajo auditoría regulatoria. En el mercado peptídico de investigación — el RUO, Research Use Only, donde la mayoría de moléculas como BPC-157, TB-500 o GHK-Cu se comercializan — ninguna autoridad obliga a esa verificación. El comprador la exige o no la exige. Quien no la exige, recibe lo que el proveedor decide entregar.
Por qué un único número no puede describir la calidad
Un único número comprime y oculta. El número 99% HPLC-UV describe qué fracción del área bajo el cromatograma corresponde al pico principal: no garantiza que ese pico sea el péptido declarado (eso es identidad), no garantiza que el vial contenga la masa anunciada de ese péptido (eso es potencia), no garantiza ausencia de microorganismos (eso es esterilidad), no garantiza ausencia de pirógenos (eso es endotoxinas). La industria peptídica RUO ha entrenado al comprador a fijarse en un solo número precisamente porque ese número por sí solo no protege. Reducir la calidad a una cifra es un acto de marketing, no de criterio.
La diferencia entre péptido grado farmacéutico, grado clínico y RUO
Péptido grado farmacéutico (pharmaceutical grade, GMP). Fabricado bajo cGMP, con dossier de validación de método, criterios de aceptación declarados, registro lote a lote, trazabilidad de materias primas, fill-finish aséptico validado y liberación por persona cualificada. Se comercializa con autorización de mercado (FDA, EMA, AEMPS u otra agencia) o como API para fabricación bajo prescripción. Ejemplos: semaglutida Ozempic, tirzepatida Mounjaro, teduglutida Revestive.
Péptido grado clínico o compounding. Fabricado para formulación magistral por farmacias 503A o 503B en USA, o equivalentes nacionales en otros países. Calidad de materia prima cercana a GMP pero la operación de llenado final puede no estar al mismo nivel de control. La diferencia con grado farmacéutico no es la molécula sino el sistema de calidad alrededor.
Péptido RUO (Research Use Only). Comercializado explícitamente para investigación, no para uso humano. Sin auditoría regulatoria del proceso. Sin obligación de verificar identidad por LC-MS, esterilidad por USP <71> o endotoxinas por USP <85>. La etiqueta RUO no protege a nadie; describe simplemente el marco legal bajo el que el producto se vende. La mayor parte del mercado de péptidos no aprobados opera aquí.
Las cuatro dimensiones que componen la calidad de un péptido
1. Pureza química — el porcentaje de la sustancia que es lo declarado
La pureza química responde a la pregunta «¿qué fracción del material analizado corresponde a la molécula objetivo y qué fracción son impurezas detectables?». Se mide por separación cromatográfica (HPLC-UV principalmente, complementada con LC-MS) y se expresa en porcentaje de área bajo el cromatograma. El estándar farmacéutico habitual es ≥ 98% de pureza para producto peptídico terminado destinado a uso humano. Por debajo de 95% se entra en territorio donde las impurezas individuales empiezan a exigir caracterización estructural específica.
2. Identidad — confirmar que es ese péptido y no otro parecido
La identidad responde a «¿la molécula mayoritaria del vial es exactamente el péptido que la etiqueta declara, o es un análogo, un isómero o un truncamiento de masa similar?». Se verifica por espectrometría de masas (LC-MS, HRMS), por análisis de aminoácidos (AAA), por mapeo peptídico con digestión enzimática, y en casos complejos por RMN. La identidad es el dato crítico que distingue péptidos cuya masa es idéntica pero cuya secuencia, topología o estereoquímica difiere — un escenario difícil de imaginar pero documentado en casos como el aducto puente metileno-tioacetal de eloralintide.
3. Esterilidad — ausencia de microorganismos viables
La esterilidad responde a «¿el contenido del vial está libre de bacterias, hongos y levaduras viables capaces de proliferar tras la inyección?». Se verifica por cultivo según USP <71> Sterility Tests o el equivalente europeo Ph. Eur. 2.6.1. Es una propiedad binaria a efectos de aceptación del lote: pasa o no pasa. La esterilidad no se mide en un cromatograma. Un péptido al 99,9% de pureza cromatográfica puede ser no estéril si el llenado aséptico falló.
4. Endotoxinas — ausencia de pirógenos bacterianos
Las endotoxinas son lipopolisacáridos (LPS) liberados por bacterias gramnegativas al lisarse. Aunque la bacteria esté muerta y por tanto la muestra sea estéril, el LPS persiste y es altamente pirógeno por activación del receptor TLR4. Se cuantifica por USP <85> Bacterial Endotoxins Test (BET) en sus variantes LAL o, más modernamente, Recombinant Factor C (rFC). El límite farmacopéico estándar para parenterales es 5 EU/kg/hora, donde EU es Endotoxin Unit. Por encima de ese límite el riesgo de pirexia, taquicardia, hipotensión e inflamación sistémica es real.
La regla KRECE: cuatro dimensiones por lote, ninguna negociable
Las cuatro dimensiones son independientes en sentido técnico — verifican propiedades distintas con métodos distintos — pero indisociables en sentido clínico: un péptido que falle cualquiera de las cuatro no es seguro inyectarlo. KRECE no acepta un proveedor que solo entregue HPLC-UV por lote. Tampoco acepta «hicimos esterilidad y endotoxinas en el primer lote y desde entonces confiamos». Lote es lote: cada uno se verifica.
| Dimensión | Pregunta que responde | Método estándar | Especificación |
|---|---|---|---|
| Pureza | ¿Qué fracción del material es la molécula objetivo? | HPLC-UV + LC-MS | ≥ 98% área |
| Identidad | ¿Es exactamente este péptido? | LC-MS / HRMS, AAA, peptide mapping | Masa exacta < 5 ppm error |
| Esterilidad | ¿Hay microorganismos viables? | USP <71> / Ph. Eur. 2.6.1 | Pasa / No pasa |
| Endotoxinas | ¿Hay LPS bacteriano? | USP <85> (LAL o rFC) | < 5 EU/kg/hora parenteral |
Por qué la matriz importa. Cada vez que un proveedor entrega «solo HPLC-UV» está trasladando al médico la responsabilidad de tres pruebas que no se han hecho. El médico raramente tiene la infraestructura para verificarlas. El paciente queda expuesto a riesgos que el cromatograma no podía detectar. La cuestión no es si el proveedor es honesto — muchos lo son. La cuestión es si el sistema de calidad genera la información necesaria. Sin las cuatro dimensiones, no la genera.
Cómo se mide la pureza de un péptido
HPLC-UV (cromatografía líquida de alta resolución con detector UV)
HPLC-UV es el caballo de batalla de cualquier laboratorio analítico peptídico. El principio es separar las moléculas de una mezcla según su afinidad por una fase estacionaria (típicamente C18 en cromatografía de fase reversa), eluirlas con un gradiente de solvente y detectarlas por absorción ultravioleta. Para péptidos la longitud de onda habitual es 214 nm — donde absorbe el enlace peptídico — o 220 nm. El resultado es un cromatograma donde cada compuesto aparece como un pico a su tiempo de retención característico, y la pureza se calcula como el porcentaje del área del pico principal sobre el área total integrada.
Por qué la pureza HPLC-UV no es la pureza del vial
Aquí está el punto crítico que la mayoría del marketing peptídico RUO esconde. El número 99% HPLC-UV es 99% del área bajo el cromatograma UV a esa longitud de onda, no 99% del contenido del vial. Impurezas que no absorben a 214 nm — sales, contraiones como el TFA, agua residual, excipientes — no aparecen en el cromatograma. Picos por debajo del umbral de integración (típicamente 0,05% del área total) se excluyen automáticamente. Y moléculas que coeluyen con el pico principal — tiempos de retención idénticos o casi idénticos — se cuentan dentro del 99%.
LC-MS — espectrometría de masas acoplada a cromatografía
LC-MS resuelve dos de las tres limitaciones del HPLC-UV. La separación cromatográfica es la misma, pero la detección no se basa en absorción UV sino en la relación masa-carga (m/z) de cada molécula tras ionización. Esto permite (a) detectar compuestos que no absorben a 214 nm pero sí ionizan, y (b) distinguir coelusiones cromatográficas si las masas de los compuestos coeluidos son diferentes. Para un péptido, LC-MS no solo cuantifica sino que confirma la identidad de cada pico por su masa característica.
HRMS — espectrometría de masas de alta resolución
HRMS (High-Resolution Mass Spectrometry, típicamente con equipos Q-TOF u Orbitrap) lleva la sensibilidad un paso más allá. Mide la masa con error inferior a 5 ppm. Esto permite confirmar la fórmula molecular exacta del péptido y detectar variantes que un LC-MS de baja resolución no separaría. Es el método de elección para detectar deamidaciones, oxidaciones puntuales y otras modificaciones post-traduccionales que cambian la masa en unidades discretas.
Análisis de aminoácidos (AAA) como método cuantitativo confirmatorio
El análisis de aminoácidos (AAA, Amino Acid Analysis) es una técnica complementaria fundamental que la mayoría de proveedores RUO no entrega. Consiste en hidrolizar el péptido en sus aminoácidos individuales mediante hidrólisis ácida (HCl 6 N, 110 °C, 18-24 h o equivalente), cuantificar cada aminoácido por cromatografía de intercambio iónico con derivatización post-columna, y comparar la composición molar observada con la teórica de la secuencia.
El AAA responde a una pregunta que HPLC-UV no puede responder: cuánto péptido real hay en el vial (el llamado Net Peptide Content o assay), independientemente de cuánta sal, agua o contraión acompañan a la molécula. Es el método de referencia de la USP <1057> para determinar potencia peptídica por vía no biológica. Su omisión sistemática en los certificados de análisis del mercado RUO no es accidental: declarar un assay del 75% «suena menos puro» al comprador no técnico, aunque sea exactamente el dato que el médico necesita para dosificar correctamente.
Espectrometría de masas en tándem (MS/MS) y secuenciación
MS/MS fragmenta el ion peptídico dentro del espectrómetro mediante colisión con un gas inerte, generando iones hijos cuyas masas mapean la secuencia de aminoácidos. Combinada con búsqueda en bases de datos (bottom-up) o secuenciación directa (top-down y de novo), confirma no solo la masa total sino el orden exacto de los aminoácidos. En péptidos cortos (< 30 aminoácidos) la secuenciación top-down por colisión inducida (CID) o disociación por transferencia electrónica (ETD) suele ser definitiva.
¿Cuál es el método de referencia para purity testing?
No hay un único método de referencia universal. La práctica farmacéutica estándar combina al menos dos métodos ortogonales: HPLC-UV para cuantificar pureza cromatográfica, LC-MS para confirmar identidad y detectar impurezas no UV-activas, y AAA o equivalente para determinar el assay real del vial. Para péptidos novedosos o estructuralmente complejos, la USP <1057> recomienda añadir MS/MS de identidad y, en casos específicos, RMN para verificar conformación. Cualquier proveedor que solo entregue HPLC-UV está entregando un subconjunto del análisis mínimo aceptable.
Identidad — verificar que es el péptido correcto, no uno parecido
Masa molecular exacta y error de masa (ppm)
El primer test de identidad es comparar la masa molecular monoisotópica observada con la teórica calculada desde la secuencia. Un espectrómetro de masas de baja resolución alcanza error de masa de aproximadamente 100-500 ppm; uno de alta resolución (HRMS, Q-TOF u Orbitrap) baja a < 5 ppm. El límite habitual de aceptación para confirmación de identidad es error de masa < 5 ppm para HRMS. Por encima de ese umbral, la identidad no se da por confirmada y se exige técnica adicional.
Mapeo peptídico (peptide mapping) y digestión enzimática
El mapeo peptídico consiste en digerir el péptido con una proteasa de especificidad conocida — típicamente tripsina (corta tras Lys y Arg), Glu-C (corta tras Glu) o quimotripsina — y analizar los fragmentos por LC-MS. El patrón de fragmentos resultante es una huella dactilar única que se compara con el patrón teórico de la secuencia declarada. El mapeo detecta sustituciones puntuales de aminoácidos que en la masa total serían indetectables por compensación. Para péptidos de cadena larga (> 40 aminoácidos) o productos biológicos, es una prueba habitualmente requerida bajo Ph. Eur. 2.2.56 y caracterización inicial ICH Q6B.
Resonancia magnética nuclear (RMN/NMR) y estructura tridimensional
La RMN, típicamente 1H o experimentos 2D (COSY, NOESY, TOCSY, HSQC), aporta información estructural que ningún otro método entrega: la conectividad covalente exacta y la conformación tridimensional. Es el método de referencia para confirmar puentes disulfuro, enlaces lactama cíclicos, ciclaciones cabeza-cola y, en general, la topología 3D del péptido. Es técnicamente cara, requiere muestra concentrada (decenas de miligramos en muchos casos) e interpretación experta, por lo que no se aplica rutinariamente por lote sino en caracterización inicial de un péptido novedoso según las guías ICH Q6B. Su importancia específica en péptidos cíclicos se desarrolla en el artículo dedicado a la topología y verificación de péptidos cíclicos.
Aductos, isómeros y «lookalikes»: cuando dos péptidos pesan casi igual
La trampa analítica más desagradable es el conjunto de moléculas que comparten masa pero no estructura. Un dipéptido modificado puede tener la misma masa nominal que otro dipéptido con secuencia invertida. Una racemización (cambio de un L-aminoácido a su forma D) no cambia la masa pero altera por completo la afinidad receptora. Un aducto covalente puede añadir una masa pequeña que un MS de baja resolución no separa del pico principal.
Secuenciación de novo por MS/MS
La secuenciación de novo intenta determinar la secuencia completa sin asumir conocimiento previo. Es la técnica de elección cuando se sospecha que el péptido recibido no es el declarado — por ejemplo, en muestras de mercado gris donde la confianza en la etiqueta es nula. Combina top-down (fragmentación del péptido intacto) y bottom-up (fragmentación de productos de digestión) según el tamaño de la molécula. En auditorías forenses publicadas del mercado peptídico no regulado se han documentado viales etiquetados como un péptido de 15 aminoácidos que contenían en realidad una secuencia de 13 aminoácidos con dos deleciones — truncamientos farmacológicamente activos pero biológicamente distintos del producto declarado.
Impurezas relacionadas con el producto — qué son y por qué importan
Las impurezas peptídicas se dividen en dos grandes familias: impurezas relacionadas con el proceso (residuos de reactivos, solventes, contraiones, fragmentos de resina) y impurezas relacionadas con el producto (variantes químicas del péptido objetivo, generadas durante síntesis o degradación post-síntesis). Las segundas son las que más riesgo clínico introducen porque comparten estructura básica con el péptido activo y pueden tener actividad farmacológica propia — agonismo parcial, antagonismo, inmunogenicidad. Las guías ICH Q3A(R2) y Q3B(R2) establecen los umbrales generales aplicables: reporte 0,05%, identificación 0,10%, calificación 0,15%.
Deamidación (Asn → Asp/isoAsp, Gln → Glu)
La deamidación es la conversión no enzimática del grupo amida lateral de la asparagina (Asn) o la glutamina (Gln) en su correspondiente ácido (Asp o Glu). En el caso de Asn, el mecanismo pasa por un intermediario succinimida que puede abrirse hacia Asp o hacia isoaspartato (isoAsp), una molécula con la cadena principal modificada. Es la vía de degradación más frecuente en solución acuosa, acelerada por pH alcalino (> 7), temperatura elevada y humedad residual elevada en producto liofilizado. Aunque la masa del péptido cambia poco (1 Da perdido por cada deamidación), la actividad biológica puede caer significativamente al alterarse la conformación local.
Oxidación (Met, Trp, Cys)
La oxidación afecta principalmente a tres aminoácidos: metionina (Met → metionina sulfóxido), triptófano (Trp → oxindolilalanina, kinurenina) y cisteína (Cys → sulfínico, sulfónico). Es muy rápida en solución cuando hay oxígeno disuelto, especies reactivas de oxígeno (ROS) o luz UV. La oxidación de Met se documenta como mecanismo de pérdida de afinidad receptora en secretagogos de hormona de crecimiento como CJC-1295 o sermorelina. Detectable por LC-MS por el shift característico de +16 Da por átomo de oxígeno incorporado.
Racemización (formación de D-aminoácidos)
La racemización es la inversión de la configuración quiral del carbono alfa, convirtiendo un L-aminoácido en su correspondiente D-aminoácido. Se favorece especialmente en cisteína, histidina y aspartato durante el acoplamiento en síntesis en fase sólida (SPPS), cuando los reactivos de activación son agresivos o el tiempo de acoplamiento se prolonga. La racemización es invisible al LC-MS: la masa de un D-aminoácido es idéntica a la de su L-isómero. Se detecta por cromatografía quiral, AAA con derivatización quiral, o RMN. Un único D-aminoácido en una secuencia que debería ser todo-L puede invalidar la actividad biológica del péptido.
Truncamientos de secuencia (deleciones n-1, n-2)
Los truncamientos aparecen cuando un ciclo de acoplamiento en SPPS no acopla completamente el siguiente aminoácido y la síntesis continúa sobre la cadena incompleta. El resultado es una mezcla del péptido completo de longitud n con variantes n-1, n-2 o más acortadas. Lo que hace que estos truncamientos sean clínicamente relevantes es que pueden actuar como agonistas parciales o antagonistas competitivos en el mismo receptor que el péptido completo, distorsionando la respuesta dosis-efecto.
Acetilación, aductos y modificaciones post-síntesis
Las modificaciones laterales no buscadas son variadas: acetilación N-terminal residual de la protección SPPS, formación de aductos con formaldehído del proceso, aductos covalentes con excipientes o con la propia molécula del péptido formando dímeros. El aducto tirzepatida-B12 documentado por Eli Lilly es un caso reciente y cuantitativamente importante: hasta el 10% del contenido peptídico de muestras de tirzepatida compounded se observó como aducto covalente con vitamina B12 añadida intencionalmente como «estabilizante» en algunas formulaciones magistrales.
TFA residual y contraiones — el dato que casi nadie declara
El ácido trifluoroacético (TFA) se usa como modificador iónico en la purificación por RP-HPLC preparativa. Al ser la fase reversa ácida, el péptido cargado positivamente forma pares iónicos con el TFA y termina liofilizado con TFA como contraión. La masa de TFA residual puede representar entre el 10% y el 30% del peso seco del vial según la cantidad de residuos básicos (Lys, Arg, His) en la secuencia y la eficiencia del intercambio de contraión previo a la liofilización.
El impacto clínico es directo: si un vial declara «5 mg de péptido» sin especificar si esa cifra es peso bruto o assay neto, el médico puede estar administrando un 20-25% menos de péptido activo del prescrito. Además, el TFA residual a concentraciones elevadas es citotóxico en cultivos celulares e introduce variabilidad farmacocinética. La práctica farmacéutica estándar exige declarar el contraión y, cuando es posible, intercambiarlo por acetato (menos tóxico) o cloruro. La práctica del mercado RUO es no declararlo.
Por qué un péptido al 99% HPLC-UV puede contener 10-15% de impurezas reales
Combinando los puntos anteriores se entiende la aparente paradoja. Una muestra puede arrojar 99,72% de pureza HPLC-UV y, al analizarse por LC-MS de alta resolución, mostrar 13,3% de impurezas reales: 6% de acetilación lateral, 7% de deamidación y picos diminutos por debajo del umbral de integración del HPLC-UV. El caso documentado de SS-31 publicado en KRECE detalla exactamente esta dinámica: las impurezas relacionadas con el producto se ocultan en el ruido del HPLC-UV estándar. Su detección requiere el combinado HPLC-UV + LC-MS aplicado simultáneamente al mismo lote.
| Impureza | Origen | Detección | Umbral ICH Q3A/Q3B |
|---|---|---|---|
| Deamidación | Degradación acuosa, pH alto | LC-MS (+1 Da), HPLC | Calificación > 0,15% |
| Oxidación Met/Trp | Oxígeno, luz UV | LC-MS (+16, +32 Da) | Calificación > 0,15% |
| Racemización | Síntesis SPPS agresiva | Cromatografía quiral, RMN | Caso por caso |
| Truncamiento n-1 | Acoplamiento SPPS incompleto | HPLC + LC-MS | Identificación > 0,10% |
| Acetilación lateral | Protección SPPS residual | LC-MS (+42 Da) | Identificación > 0,10% |
| TFA residual | Purificación RP-HPLC | NMR 19F, IC, Karl Fischer | Específico de monografía |
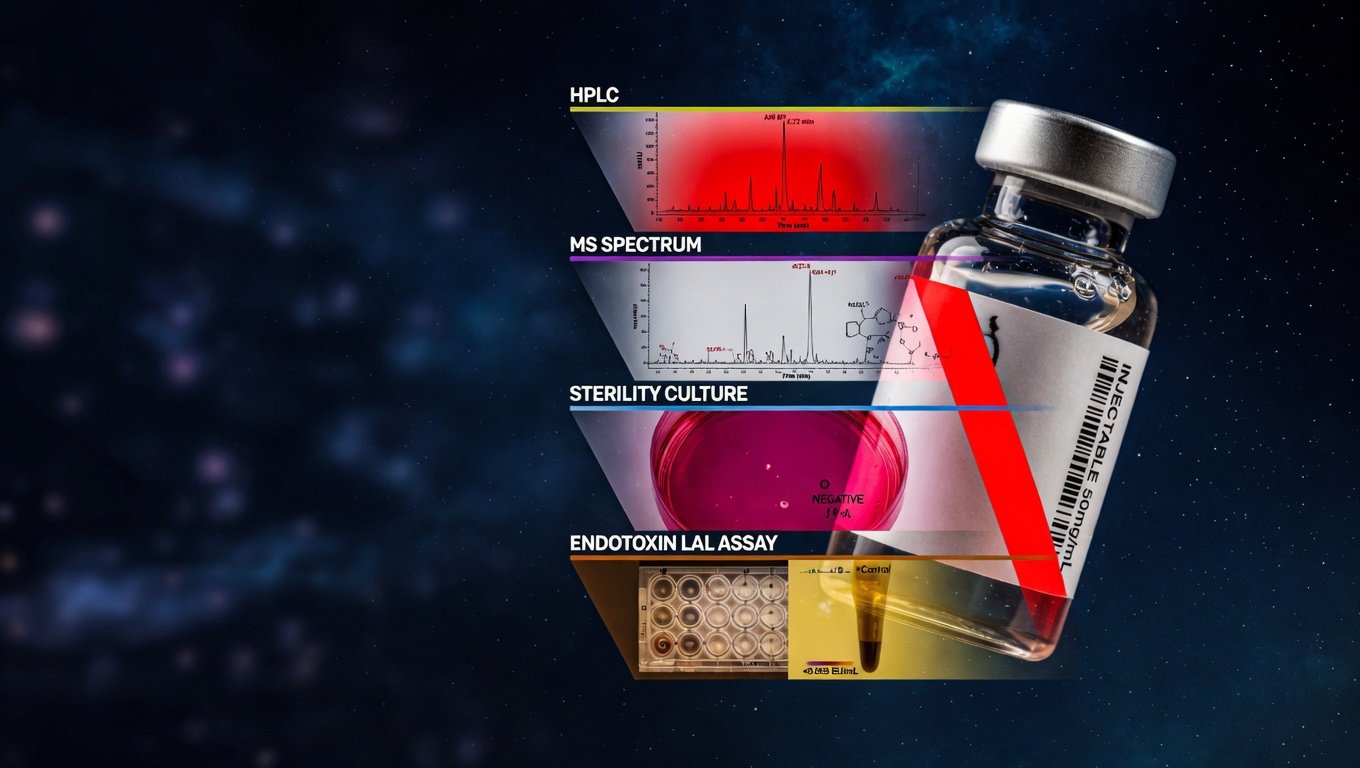
Un certificado de análisis aceptable declara explícitamente: pureza HPLC-UV (% área) y pureza LC-MS (% por masa), assay por AAA o método equivalente, identidad confirmada por masa exacta con error en ppm, contraión declarado, humedad residual por Karl Fischer, esterilidad según USP <71> y endotoxinas según USP <85>. Si falta cualquiera de estos siete puntos, el CoA es incompleto.
Esterilidad — qué garantiza, qué no garantiza
USP <71> Sterility Tests — método oficial estadounidense
La USP <71> define el método compendial para verificar la ausencia de microorganismos viables en productos farmacéuticos estériles. El procedimiento estándar es la filtración por membrana: el contenido del vial se filtra a través de una membrana de 0,45 µm que retiene microorganismos; la membrana se transfiere a dos medios de cultivo (caldo tioglicolato fluido para anaerobios y bacterias aerobias, y caldo de soja-caseína para hongos y aerobios), se incuba 14 días a temperaturas controladas, y se inspecciona visualmente la turbidez. Pasa si no hay crecimiento. Falla si hay crecimiento de cualquier tipo, en cualquier tubo.
Ph. Eur. 2.6.1 — equivalente europeo
La Farmacopea Europea recoge en el capítulo 2.6.1 un método armonizado con la USP <71> y con la Farmacopea Japonesa (JP 4.06). En la práctica industrial son intercambiables: un lote que pase USP <71> pasa Ph. Eur. 2.6.1, y viceversa, en condiciones normales. La diferencia es jurisdiccional, no técnica. Para producto destinado a mercado europeo, se referencia Ph. Eur.; para USA, USP; para registro multinacional, ambas.
Microorganismos detectados típicamente
Cuando un lote falla esterilidad, el siguiente paso analítico es identificar el microorganismo. La técnica habitual es MALDI-TOF (espectrometría de masas de tiempo de vuelo con desorción láser asistida por matriz) aplicada al cultivo positivo. Las identificaciones típicas en lotes peptídicos RUO incluyen flora ambiental común — Staphylococcus epidermidis, Micrococcus luteus, Bacillus spp., Aspergillus spp. — pero también especies de relevancia clínica directa — Staphylococcus aureus, Pseudomonas aeruginosa, Enterococcus — cuya presencia en producto inyectable es directamente perjudicial.
Esterilidad no es lo mismo que apirógeno
Este es un punto que se confunde regularmente. Esterilidad mide microorganismos viables: capaces de proliferar tras inocularse en medio. Una bacteria muerta por autoclave o por irradiación no es viable, pero su esqueleto puede contener endotoxinas activas. Un lote puede pasar USP <71> (todos los microorganismos están muertos) y fallar USP <85> (las endotoxinas residuales son pirogénicas). Por eso ambas pruebas son obligatorias y no se sustituyen una a la otra. Un CoA que declare solo esterilidad sin endotoxinas, o solo endotoxinas sin esterilidad, está incompleto.
Endotoxinas — el riesgo invisible que no aparece en el cromatograma
Qué es una endotoxina (lipopolisacárido bacteriano)
Las endotoxinas son lipopolisacáridos (LPS) que forman parte de la membrana externa de las bacterias gramnegativas. Cuando estas bacterias mueren, los LPS se liberan al medio. Son térmicamente estables (sobreviven autoclave a 121 °C), resisten filtración por 0,22 µm y no se inactivan por la mayoría de tratamientos estándar. En cuanto un LPS entra en circulación humana activa receptores TLR4 en macrófagos y células endoteliales, desencadenando una cascada de citoquinas proinflamatorias (TNFα, IL-1, IL-6) cuya manifestación clínica es pirexia, taquicardia, hipotensión y, a dosis altas, shock séptico.
USP <85> Bacterial Endotoxins Test — LAL clásico
El método histórico estándar es el LAL (Limulus Amebocyte Lysate), basado en la cascada enzimática natural de los amebocitos del cangrejo herradura Limulus polyphemus. En presencia de LPS la cascada se activa y produce una señal detectable. Existen tres variantes operativas: gel-clot (cualitativo, pasa/no pasa por formación de gel), turbidimétrico cinético (cuantitativo, mide opacidad creciente) y cromogénico cinético (cuantitativo, usa sustrato sintético que libera cromóforo). El cromogénico cinético es el más usado en industria por su rango dinámico y precisión.
Recombinant Factor C (rFC) — alternativa sin cangrejos herradura
El rFC reproduce sintéticamente el sensor de endotoxinas del cangrejo — el Factor C — sin necesidad de extraer hemolinfa del animal. Es bioquímicamente específico por LPS (el LAL natural puede dar falsos positivos por β-glucanos que activan el Factor G de la cascada), reproducible lote a lote, éticamente preferible y sostenible. La Farmacopea Europea fue pionera en codificarlo, y el capítulo Ph. Eur. 5.1.10 recomienda activamente la transición a rFC. La USP, más conservadora, consolidó en 2024 el capítulo USP <86> para métodos recombinantes equivalentes, otorgándoles estatus compendial.
Límite farmacopéico: 5 EU/kg/hora para parenterales
El límite estándar para productos parenterales no intratecales es 5 EU/kg/hora, donde EU (Endotoxin Unit) es la unidad farmacopéica. Para un adulto de 70 kg, esto se traduce en un límite total absoluto de 350 EU por hora. Si el péptido se administra como dosis única de 5 mg, el límite máximo de endotoxinas por miligramo es 70 EU/mg. Para productos intratecales el límite cae a 0,2 EU/kg, mucho más estricto. Estos límites se ajustan según la dosis, frecuencia de administración y vía.
Por qué un péptido «puro» puede provocar fiebre
El cromatograma no ve LPS. La esterilidad no ve LPS muerto. Un lote puede atravesar limpiamente HPLC-UV, LC-MS y USP <71> y aún así contener cantidades pirogénicas de endotoxinas. La inyección provoca fiebre, escalofríos, taquicardia y, en cuadros más severos, hipotensión. En el reporte Qualsera PQI 2026 sobre 423 muestras peptídicas analizadas por USP <85>, 76 muestras (18%) superaron el límite de cuantificación — es decir, contenían endotoxinas detectables. La mayoría por debajo del límite clínico, pero la presencia en sí ya señala fallo del sistema de calidad.
Potencia — ¿el vial contiene realmente la dosis que declara?
Label claim y la regla del ±10%
La potencia es la fracción del vial que efectivamente contiene la molécula activa, expresada como porcentaje del label claim (la cifra que aparece en la etiqueta). El estándar farmacéutico en USA y UE es 90-110% del label claim — un vial etiquetado como 5 mg debe contener entre 4,5 y 5,5 mg de péptido activo para considerarse dentro de especificación. Esta tolerancia del ±10% es generosa: refleja la dificultad operativa de llenado aséptico de viales individuales, no una imprecisión clínica aceptable.
Sub-dosing y over-dosing: por qué los dos son problemas distintos
El sub-dosing (por debajo del 90%) implica que el paciente recibe menos fármaco del prescrito y, en consecuencia, menos efecto terapéutico. Para un agonista GLP-1 prescrito a dosis clínica, un vial al 75% del label claim significa que el paciente está recibiendo una dosis terapéuticamente subóptima sin saberlo. El over-dosing (por encima del 110%) es menos intuitivamente problemático pero igualmente serio: a más péptido por vial, más efectos adversos por dosis, y en péptidos con ventana terapéutica estrecha, mayor riesgo de toxicidad. Ambos rompen la relación entre prescripción y exposición real.
Cómo se mide la potencia en un péptido
Para péptidos sintéticos el método más riguroso es el AAA (análisis de aminoácidos), que cuantifica la masa real de péptido en el vial independiente de contraiones, sales y agua residual. Métodos secundarios incluyen HPLC con estándar externo cuantitativo, análisis por nitrógeno total (Kjeldahl o combustión), o ensayos biológicos específicos para péptidos con receptor identificado (habitual en farmacéutico, raro en RUO). El error frecuente del mercado peptídico no regulado es declarar como potencia el peso bruto del vial, que incluye TFA, agua y posibles excipientes — sobreestimando sistemáticamente la dosis activa.
Cuando el vial dice «5 mg» y contiene 0 mg: viales sin péptido en el mercado
El extremo cuantitativo del fallo de potencia es el vial sin péptido alguno. El reporte Qualsera PQI 2026 documentó explícitamente la existencia de viales etiquetados como GLP-1 que contenían 0% del label claim — es decir, no contenían el péptido declarado en absoluto. El mismo patrón se observó en viales etiquetados como BPC-157 y TB-500. No se trata de variabilidad de proceso: se trata de viales que probablemente nunca recibieron péptido durante el fill-finish, o que recibieron una sustancia distinta. Cualquiera de las dos opciones es un fallo total del sistema de trazabilidad de lote.
Datos reales del mercado de péptidos en 2025-2026
El reporte Qualsera Peptide Quality Index 2026 analizó entre 70 y 837 datos de potencia por molécula procedentes de testeo independiente de productos peptídicos del mercado durante el año 2025. El reporte clasifica internamente la tirzepatida bajo la etiqueta abreviada «GLP-3» para simplificar nomenclatura; estrictamente es un co-agonista dual GIP/GLP-1, no un «GLP-3» biológico (entidad que no existe). Los siguientes apartados resumen los hallazgos por molécula y la tendencia 2024 → 2025. La cifra titular en cada caso es el porcentaje de viales dentro de especificación de potencia (90-110% del label claim) o de pureza (≥ 98% en HPLC).
26% de viales GLP-1 dentro de especificación de potencia
Sobre 289 datos de potencia de GLP-1 analizados en 2025, el promedio fue 116,8% del label claim y solo el 26% de los viales estaba dentro del rango 90-110%. En 2024 ese porcentaje era 40%. La tendencia es de deterioro claro: a medida que el mercado se ha expandido por la escasez de Ozempic y Wegovy y la demanda compounded ha crecido, la consistencia ha empeorado. En pureza, 94,5% de las muestras estaba ≥ 98%, pero los valores bajos se asociaban a componentes no identificados no resueltos por análisis estándar.
19% de viales de tirzepatida (co-agonista GIP/GLP-1) dentro de especificación — el peor del estudio
Tirzepatida es la molécula con peor consistencia de potencia del reporte: sobre 610 datos analizados, promedio del 115,1% del label claim, y solo el 18,7% dentro de especificación (vs 20,7% en 2024). El sesgo es sistemáticamente al alza — es decir, los viales contienen más péptido del declarado, probablemente como decisión comercial para reducir riesgo de underfill. Pero el efecto sobre el paciente es una exposición consistentemente superior a la prescrita. En pureza, 92,2% de las muestras ≥ 98%, con datos clustered entre 90-98% que sugieren degradación por liofilización incompleta o estabilidad insuficiente.
27% de viales TB-500 dentro de especificación
Para TB-500 se analizaron 117 datos: promedio 90,8% del label claim, 27,4% dentro de especificación. Aquí el sesgo va al revés, hacia el underfill: la mayoría de los viales contienen menos péptido del declarado. En pureza, 84,3% de las muestras ≥ 98% — el valor más bajo del reporte, asociado nuevamente a componentes no resueltos en HPLC estándar.
35% de viales BPC-157 dentro de especificación
BPC-157 se analizó sobre 260 datos: promedio 106,6% del label claim (subiendo desde 97,2% en 2024), 35,4% dentro de especificación (cayendo desde 44,4% en 2024). El reporte documentó explícitamente la existencia de viales etiquetados como BPC-157 que no contenían BPC-157 alguno. Para una molécula que es el caballo de batalla de la promesa peptídica en reparación tisular, el dato es severo.
12,7% de muestras no estériles documentadas
Sobre 433 muestras analizadas para esterilidad según USP <71>, 55 muestras (12,7%) fueron no estériles. La identificación por MALDI-TOF reveló una mezcla de organismos ambientales comunes y organismos de potencial relevancia clínica. El dato 12,7% no significa que el 12,7% del mercado total esté contaminado — el dataset Qualsera incluye muestras enviadas para verificación por sospecha, no muestreo aleatorio. Pero la cifra describe la magnitud del fallo en el subconjunto que llega a verificación.
18% de muestras con endotoxinas por encima del LOQ
Sobre 423 muestras analizadas para endotoxinas según USP <85>, 76 muestras (18%) contenían endotoxinas por encima del límite de cuantificación. La mayoría por debajo del límite clínico de 5 EU/kg/hora, pero la presencia de endotoxinas detectables ya señala fallo del control de procesos asépticos. En inyecciones diarias durante semanas o meses la exposición acumulada puede convertirse en relevante.
La tendencia 2024 → 2025 — la consistencia está empeorando, no mejorando
El patrón agregado más preocupante del reporte Qualsera es la trayectoria interanual. En todas las moléculas analizadas con comparativa 2024 vs 2025 (GLP-1, GLP-2, tirzepatida, BPC-157), el porcentaje de viales dentro de especificación descendió. GLP-1 pasó de 40% a 26%. GLP-2 de 50% a 41,7%. BPC-157 de 44,4% a 35,4%. El crecimiento del mercado está ocurriendo más deprisa que la maduración del sistema de calidad de los fabricantes que entran a operar en él. La calidad media del mercado peptídico no regulado está empeorando en 2025, no mejorando.
| Molécula | n (potencia) | Promedio % LC | % in spec 2025 | vs 2024 |
|---|---|---|---|---|
| GLP-1 (semaglutida) | 289 | 116,8% | 26,0% | ↓ desde 40,0% |
| GLP-2 (teduglutida) | 837 | 108,6% | 41,7% | ↓ desde 50,0% |
| Tirzepatida (GIP/GLP-1) | 610 | 115,1% | 18,7% | ↓ desde 20,7% |
| BPC-157 | 260 | 106,6% | 35,4% | ↓ desde 44,4% |
| TB-500 | 117 | 90,8% | 27,4% | sin comparativa |
El Certificado de Análisis (CoA) — cómo se lee y qué busca el ojo entrenado
Información obligatoria — lote, fecha, métodos, criterios de aceptación
Un CoA mínimamente aceptable identifica unívocamente el material analizado y el sistema que lo produjo. Debe contener: nombre y secuencia del péptido, número de lote y fecha de fabricación, fecha de análisis, fabricante y laboratorio analítico (si son distintos), métodos analíticos usados con referencia compendial (USP/Ph. Eur.) o protocolo interno declarado, criterios de aceptación para cada parámetro, resultados obtenidos para cada parámetro, y firma o identificación del responsable de liberación del lote.
Las cuatro secciones imprescindibles de un CoA completo
El CoA completo refleja la matriz de las cuatro dimensiones: (1) Pureza, declarando porcentaje HPLC-UV con cromatograma adjunto o referenciado, y porcentaje LC-MS si se realizó ortogonalmente; (2) Identidad, declarando masa exacta observada vs teórica con error en ppm y el espectro de masas adjunto; (3) Esterilidad, declarando método USP <71> o Ph. Eur. 2.6.1 con resultado pasa/no pasa; (4) Endotoxinas, declarando método USP <85> con valor en EU/mg y comparación contra el límite calculado para la indicación. Adicionalmente: assay por AAA o equivalente (con su % de Net Peptide Content), humedad residual por Karl Fischer, y contraión declarado.
Señales de alarma en un CoA
Las omisiones más frecuentes en CoAs de mercado RUO son: ausencia de LC-MS (solo HPLC-UV); ausencia de assay (solo pureza cromatográfica); ausencia de esterilidad o endotoxinas (a veces ambas); valores genéricos sin número de lote único (sospechoso de plantilla reutilizada); ausencia de cromatograma adjunto; ausencia de firma o identificación del responsable. Cualquiera de estas faltas degrada el CoA a marketing material. La práctica KRECE es solicitar el CoA antes de comprar y rechazar inmediatamente los que omiten cualquiera de las cuatro dimensiones.
Cuando un CoA «bonito» oculta un péptido defectuoso
El caso más instructivo en la literatura KRECE es el del péptido SS-31, donde un CoA declaraba 99,72% de pureza HPLC-UV y, en análisis ortogonal por LC-MS, reveló 13,3% de impurezas reales no separables en el HPLC estándar. Cubrimos el caso completo en el análisis del caso SS-31. El patrón se repite: un CoA con un solo método analítico es bonito pero ciego. El comprador que no exige métodos ortogonales está aceptando el sesgo del proveedor.
¿Sirve un CoA con solo HPLC-UV? (no)
Operativamente, no. Un CoA con únicamente HPLC-UV cubre una de las cuatro dimensiones y deja las otras tres sin verificar. Para uso humano parenteral, esto es insuficiente como cuestión de seguridad básica. Para investigación in vitro o en modelo animal puede ser tolerable según el riesgo. La regla KRECE es explícita: ningún lote entra al catálogo si el CoA no documenta las cuatro dimensiones por lote, no por proveedor.
Cadena de suministro — la calidad empieza antes del vial
Síntesis SPPS (Solid-Phase Peptide Synthesis) y sus modos de fallo
La síntesis en fase sólida es la tecnología estándar para producción de péptidos sintéticos desde los años 60. El proceso es iterativo: cada aminoácido se acopla secuencialmente sobre una resina mediante reactivos de activación (HBTU, HATU, DIC/Oxyma o similares) y se desprotege el grupo Fmoc o tBoc antes del siguiente ciclo. Los modos de fallo principales son: acoplamientos incompletos que generan truncamientos n-1; deprotecciones incompletas que generan péptidos con grupo protector residual; racemizaciones por activación agresiva; y desprendimientos prematuros de la resina. Cada uno produce una impureza específica.
Purificación industrial por RP-HPLC preparativa
Tras la síntesis SPPS, el péptido bruto se purifica mediante cromatografía en fase reversa preparativa, típicamente con columnas C18 y gradientes acuosos de acetonitrilo modificados con TFA o ácido fórmico. La pureza del producto purificado depende de la resolución cromatográfica disponible y de qué tan próximas están las impurezas al pico principal. Para impurezas que coeluyen completamente con el producto, la purificación por HPLC no las elimina — es el caso de algunos truncamientos n-1 donde el aminoácido perdido es hidrofílicamente similar.
Liofilización — el paso donde se gana o se pierde estabilidad
La liofilización (freeze-drying) convierte el péptido purificado en solución acuosa en un sólido amorfo o cristalino estable a temperatura ambiente. El proceso tiene tres fases: congelación (formación controlada de cristales de hielo y matriz amorfa), secado primario (sublimación del hielo bajo vacío), y secado secundario (desorción del agua ligada no congelada por calentamiento controlado). Los parámetros críticos son temperatura de congelación, presión de cámara durante sublimación, y humedad residual final medida por Karl Fischer (objetivo típico ≤ 2-3% para estabilidad a largo plazo).
Los excipientes habituales son lyoprotectants (trehalosa, sacarosa) que sustituyen molecularmente el agua durante la deshidratación y protegen la conformación del péptido, y bulking agents (manitol, glicina) que dan estructura al cake liofilizado para reconstitución uniforme. Un cake bien formado es un disco poroso uniforme; un cake colapsado (consecuencia de superar la temperatura crítica durante el secado primario) es un depósito vidrioso difuso indicativo de fallo de proceso.
Llenado aséptico (aseptic fill-finish) vs reconstitución casera
El llenado aséptico industrial se realiza en salas blancas grado A bajo flujo laminar, con personal en trajes estériles, equipos esterilizados y validación por media fills periódicos. El producto se filtra por 0,22 µm inmediatamente antes del llenado, se dosifica por bomba peristáltica con peso verificado, se tapona con tapón estéril y se sella con cápsula de aluminio crimped. Cualquier fallo en este proceso compromete la esterilidad del lote entero.
La reconstitución casera por el usuario final — cargar agua bacteriostática en una jeringa y mezclarla con el contenido del vial — es por definición no aséptica en un sentido farmacéutico estricto. Las buenas prácticas (hisopo de alcohol isopropílico 70%, técnica de aguja única, descarte tras uso, no agitar el vial reconstituido) mitigan el riesgo pero no lo eliminan. Cada penetración del tapón con aguja es una oportunidad de contaminación acumulativa.
Cadena de frío y estabilidad después de salir de planta
La estabilidad declarada por un fabricante (típicamente 24 meses a 2-8 °C) asume cadena de frío continua. Cuando esa cadena se rompe — transporte sin refrigeración, almacenamiento incorrecto en distribuidor, exposición a temperatura ambiente durante días — los procesos de degradación se aceleran. Un péptido sensible a deamidación puede perder porcentajes significativos de pureza en pocas semanas a temperatura ambiente. La calidad declarada en el CoA al salir de planta no se conserva automáticamente hasta el momento del uso si la logística intermedia no la protege.
Marcos regulatorios — qué controla cada autoridad
cGMP (current Good Manufacturing Practice) — el estándar farmacéutico
La práctica correcta de fabricación vigente (cGMP) es el marco regulatorio aplicable a cualquier producto medicinal terminado en USA, UE, Japón y otras jurisdicciones de farmacopea madura. Codifica requisitos sobre diseño de instalaciones, formación de personal, calidad de materias primas, validación de procesos, control de cambios, trazabilidad, almacenamiento, distribución y farmacovigilancia post-comercialización. Producto fabricado bajo cGMP está sujeto a inspección regular por la autoridad regulatoria correspondiente (FDA, EMA, AEMPS, COFEPRIS, ANMAT, INVIMA, DINAVISA). Producto fabricado fuera de cGMP no recibe esa garantía externa.
Research Use Only (RUO) — la etiqueta que no protege a nadie
El estatus RUO define un producto comercializado explícitamente para investigación in vitro o en modelos animales preclínicos, no para uso humano. Es una categoría regulatoria de baja exigencia: no requiere los controles de cGMP, no exige verificación por lote de las cuatro dimensiones de calidad, y la responsabilidad legal recae principalmente sobre el comprador. El problema operativo es que esa misma molécula RUO se comercializa de hecho para uso humano por farmacias compounding, médicos prescriptores en mercados poco regulados y usuarios autoinstruidos. La etiqueta dice una cosa, el uso real es otra. El estatus RUO no protege al usuario final en sentido clínico ni legal una vez la sustancia entra en un cuerpo humano.
Farmacopea Europea (Ph. Eur.) y monografías peptídicas
La Farmacopea Europea, editada por la EDQM (European Directorate for the Quality of Medicines), publica monografías específicas por sustancia farmacéutica que codifican identidad, pureza, ensayo y otros parámetros de calidad. Para péptidos sintéticos la monografía marco es Ph. Eur. 2034 (Péptidos sintéticos), que define atributos comunes a la familia. Capítulos generales relevantes son 2.2.56 (mapeo peptídico), 2.6.1 (esterilidad), 2.6.14 y 5.1.10 (endotoxinas, métodos compendiales y alternativos). La Ph. Eur. fue pionera en adoptar el rFC para endotoxinas antes que la USP americana.
Pharmacopoeia de Estados Unidos (USP) y capítulos clave
La USP codifica el equivalente americano. Los capítulos más relevantes para calidad peptídica son: USP <71> (esterilidad), USP <85> (endotoxinas por LAL), USP <86> (endotoxinas por métodos recombinantes equivalentes, consolidado en 2024), USP <1057> (caracterización analítica de biotécnicos incluyendo péptidos), USP <1225> (validación de métodos analíticos) y USP <788> (partículas sub-visibles). USP y Ph. Eur. mantienen un esfuerzo continuo de armonización: la mayoría de métodos compendiales son intercambiables en la práctica.
España y UE — AEMPS y EMA frente al mercado peptídico
En España, la AEMPS regula medicamentos autorizados con código nacional. Para péptidos no autorizados con indicación clínica específica (lipodistrofia, enfermedades raras), la vía legal es la Importación de Medicamentos en Situaciones Especiales — importación nominativa tramitada por el médico para un paciente concreto. Tesamorelina (Egrifta) es ejemplo paradigmático. Para péptidos sin indicación autorizada en la UE (BPC-157, TB-500, GHK-Cu inyectable), no existe vía comercial legal en España: las fórmulas magistrales requieren que el principio activo cuente con monografía en la Real Farmacopea Española o pertenezca a una lista positiva, lo que excluye a la mayoría de péptidos de uso anabólico/regenerativo. La AEMPS persigue activamente la comercialización de viales RUO bajo apariencia de reactivos cuando se promocionan para uso humano.
MERCOSUR, ANMAT, DINAVISA, INVIMA, COFEPRIS — el mosaico LATAM
En MERCOSUR el panorama es heterogéneo. Argentina (ANMAT) ha incrementado vigilancia sobre secretagogos de hormona de crecimiento en mercado fitness, restringiendo formulación compounded de moléculas sin datos de seguridad humana fuera del ámbito de investigación clínica. Paraguay (DINAVISA) mantiene el modelo más permisivo: el uso de péptidos como sermorelina, BPC-157 (en forma oral estabilizada) y combinaciones personalizadas es legal bajo receta magistral nominativa, exigiendo a la farmacia validación de identidad estructural y CoA de tercera parte por lote. Colombia (INVIMA) mantiene los péptidos inyectables como medicamentos de control con exigencia de registro sanitario para importación no nominativa. México (COFEPRIS) ha emitido alertas sobre venta de semaglutida sin registro sanitario en clínicas estéticas. El detalle nacional importa: cada jurisdicción define qué es legal, qué requiere receta, qué debe importarse nominativamente y qué está explícitamente prohibido.
503A vs 503B en USA y el aviso FDA 2026-08552
En USA, las farmacias compounding se dividen en 503A (farmacias tradicionales que componen para pacientes individuales bajo prescripción) y 503B (instalaciones de outsourcing que pueden componer en mayor escala bajo cGMP simplificado y registro federal). El aviso FDA 2026-08552 publicado en mayo de 2026 excluye semaglutida, tirzepatida y liraglutida de la lista 503B bulks, prohibiendo a las farmacias 503B componer estos GLP-1 a gran escala tras la resolución oficial de las escaseces (tirzepatida en diciembre 2024, semaglutida en febrero 2025). Las 503A pueden aún componer estos fármacos en casos justificados individualmente (dosis fuera de estándar, intolerancia documentada a excipientes comerciales), pero la era de la disponibilidad masiva de GLP-1 compounded en USA está terminando.
Mercado gris — qué es y por qué la calidad ahí es estadísticamente peor
El mercado gris es el espacio donde se vende péptido RUO directamente al consumidor final, normalmente vía web, sin prescripción, sin verificación de identidad del comprador, sin cadena de frío documentada, sin asistencia clínica. Los datos Qualsera PQI 2026 reflejan principalmente esa parte del mercado — muestras enviadas a verificación por sospecha o por comprador exigente. Las cifras de calidad son las que son: 26% de viales GLP-1 dentro de spec, 12,7% de fallos de esterilidad, 18% de muestras con endotoxinas detectables. Operar en mercado gris sin sistema de verificación personal es aceptar esas tasas como ruleta.
Cómo evaluar a un proveedor de péptidos — el método KRECE
Los cuatro documentos que se piden por lote
El método KRECE de evaluación de proveedor empieza con la solicitud de cuatro documentos específicos antes de aceptar el lote: (1) CoA completo con las cuatro dimensiones (pureza HPLC-UV + LC-MS, identidad por masa exacta, esterilidad USP <71> o equivalente, endotoxinas USP <85> o equivalente); (2) cromatograma HPLC-UV adjunto con tiempos de retención y áreas integradas declarados; (3) espectro de masas con masa monoisotópica observada y error en ppm; (4) ficha técnica del producto con secuencia, peso molecular, contraión declarado, humedad residual por Karl Fischer y assay. La práctica detallada del análisis se desarrolla en el método KRECE de evaluación de proveedor.
Las cinco preguntas que definen si un proveedor sabe lo que hace
Tras revisar la documentación, la conversación directa con el proveedor revela su grado de competencia técnica real. Cinco preguntas son diagnósticas: (1) ¿Qué método usan para confirmar identidad más allá de HPLC-UV?; (2) ¿Cómo manejan el contraión TFA y lo declaran en el CoA?; (3) ¿Cuál es la humedad residual típica de su producto liofilizado y cómo se mide?; (4) ¿Tienen población de datos históricos de esterilidad y endotoxinas por lote o solo verifican el primer lote?; (5) ¿Cuál es su política ante un fallo de spec — rework, reproceso o destrucción? Un proveedor que no puede responder con detalle técnico a las cinco preguntas no opera con sistema de calidad establecido.
Tipos de fraude documental en CoAs — qué buscar
Los CoAs fraudulentos más comunes son: plantilla reutilizada (mismos números para distintos lotes, fecha y lote intercambiados); cromatograma stock (mismo cromatograma usado para varios lotes); métodos referenciados pero sin protocolo (USP <85> declarado pero sin resultado numérico); laboratorio inventado (laboratorio analítico que no existe o es la misma entidad que el fabricante sin separación declarada); secuencia incorrecta (el peso molecular declarado no corresponde a la secuencia indicada). La verificación cruzada es siempre útil: pedir lote 1 y lote 5 del mismo proveedor y comparar cómo difieren. Lotes idénticos al detalle son sospechosos.
Por qué KRECE descarta dos de cada tres proveedores que evalúa
En procesos de evaluación recientes documentados, KRECE ha descartado aproximadamente dos de cada tres proveedores presentados a evaluación inicial. Los motivos más frecuentes son omisión de LC-MS en el CoA, incapacidad de entregar espectro de masas con error en ppm, y ausencia de assay por AAA. El caso más didáctico es el documentado en el secuestro de marca de Peptide Sciences: cuando un proveedor desaparece, lo único que sobrevive como activo del comprador es la documentación verificable por lote. KRECE no compra marcas: compra documentación.
Preguntas frecuentes sobre calidad de péptidos
¿Qué es la calidad de un péptido en una frase?
La calidad de un péptido es la combinación simultánea, verificable por lote, de pureza química ≥ 98% (HPLC + LC-MS), identidad confirmada por masa exacta (< 5 ppm error), ausencia de microorganismos viables (USP <71> pasa) y endotoxinas por debajo de 5 EU/kg/hora (USP <85>). Cuatro pruebas independientes por lote, no una.
¿Cuál es la pureza mínima aceptable de un péptido terapéutico?
El estándar farmacéutico habitual es ≥ 98% de pureza HPLC-UV con LC-MS confirmatorio. Por debajo de 95% se entra en territorio donde las impurezas individuales empiezan a exigir caracterización estructural específica según ICH Q3A/Q3B. Para productos inyectables a uso humano crónico, 99% es deseable.
¿Cómo sé si un péptido es seguro para inyectar?
Operativamente, exigiendo CoA por lote con las cuatro dimensiones documentadas: pureza, identidad, esterilidad y endotoxinas. Un «sí, es seguro» del proveedor sin documentación verificable no constituye prueba de nada. Para uso parenteral en humanos, ningún método de verificación alternativo al CoA completo por lote es aceptable.
¿Qué pruebas debe pasar un péptido antes de ser parenteral?
Como mínimo: pureza HPLC-UV ≥ 98%, identidad por LC-MS con error de masa < 5 ppm, esterilidad según USP <71> o Ph. Eur. 2.6.1, endotoxinas según USP <85> o Ph. Eur. 2.6.14 por debajo del límite clínico calculado para la indicación, y assay por AAA o equivalente declarando el Net Peptide Content. Para producto cGMP se añade humedad residual por Karl Fischer y caracterización inicial de impurezas relacionadas con producto.
¿Es lo mismo «peptide purity» que «peptide quality»?
No. Peptide purity es una de las cuatro dimensiones de calidad — concretamente, qué fracción del material es la molécula objetivo según cromatografía. Peptide quality es la matriz completa: pureza + identidad + esterilidad + endotoxinas, más assay y otras propiedades físicas. Un péptido al 99% de pureza puede tener mala calidad si falla identidad, esterilidad o endotoxinas.
¿Qué diferencia hay entre péptido grado farmacéutico y RUO?
El péptido grado farmacéutico se fabrica bajo cGMP con verificación auditada de las cuatro dimensiones por lote y autorización regulatoria de mercado. El péptido RUO se comercializa para investigación in vitro o animal sin obligación de cGMP ni de verificación compendial por lote. La molécula es la misma; el sistema de calidad alrededor no.
¿Qué hago si el proveedor se niega a enviar el cromatograma adjunto al CoA?
Rechazar el lote. El CoA sin cromatograma adjunto no es verificable: solo presenta los números resumidos que el proveedor decidió declarar, sin posibilidad de comprobar tiempos de retención, áreas integradas, picos minoritarios ni el umbral de integración aplicado. Cualquier proveedor con sistema de calidad serio entrega el cromatograma como anexo estándar. La negativa es diagnóstica.
¿Por qué dos lotes del mismo péptido pueden dar resultados distintos?
Porque cada lote es una corrida industrial independiente sometida a variación: distinto operador, distinta resina, distinta velocidad de purificación, distinto ciclo de liofilización, distinta cadena de frío post-fabricación. La consistencia interlote es un atributo del sistema de calidad del fabricante: cuanto mejor el sistema, menor la variación. Verificar por lote es la única forma de saber si la calidad del primer lote se reproduce en el décimo.
¿Cómo se compara la calidad de un péptido compounded con uno aprobado por FDA/EMA?
El péptido aprobado por FDA/EMA (Ozempic, Mounjaro, Egrifta y similares) se fabrica bajo cGMP con auditoría regulatoria continua, verificación compendial por lote y trazabilidad completa. El péptido compounded en farmacia 503A puede alcanzar calidad similar si el laboratorio aplica los mismos estándares y verifica por lote, pero la calidad media documentada del mercado compounded es significativamente inferior — con tasas de 18-26% in spec frente al ~99% esperable de producto farmacéutico aprobado.
La calidad de un péptido no es un número. Es una matriz de cuatro dimensiones que ningún método aislado puede cubrir.
Este artículo es contenido editorial. No sustituye al criterio médico individualizado. Las decisiones sobre uso de péptidos terapéuticos, fuente de adquisición, dosis y vía corresponden al médico tratante en función de la historia clínica del paciente y del marco regulatorio aplicable en cada jurisdicción. Los datos cuantitativos sobre el mercado peptídico citados proceden de fuentes analíticas independientes; la interpretación editorial es de KRECE.
- Qualsera Inc. Peptide Quality Index 2026. Análisis de potencia y pureza en muestras peptídicas del mercado durante 2024-2025. GLP-1 (n=289), GLP-2 (n=837), GLP-3/tirzepatida (n=610), BPC-157 (n=260), TB-500 (n=117). Esterilidad USP <71> (n=433). Endotoxinas USP <85> (n=423).
- United States Pharmacopeia. USP <71> Sterility Tests. Método compendial para verificación de esterilidad de productos farmacéuticos estériles.
- United States Pharmacopeia. USP <85> Bacterial Endotoxins Test. Método compendial para cuantificación de endotoxinas por LAL.
- United States Pharmacopeia. USP <86> Bacterial Endotoxins Test — Using Recombinant Reagents. Capítulo consolidado en 2024 para métodos recombinantes (rFC).
- United States Pharmacopeia. USP <1057> Biotechnology-Derived Articles — Total Protein Assay. Determinación cuantitativa de contenido peptídico neto.
- European Pharmacopoeia. Ph. Eur. 2034 — Substances for Pharmaceutical Use, Synthetic Peptides. Monografía marco para péptidos sintéticos.
- European Pharmacopoeia. Ph. Eur. 2.2.56 — Amino Acid Analysis. Método compendial AAA.
- European Pharmacopoeia. Ph. Eur. 2.6.1 — Sterility. Método compendial para esterilidad.
- European Pharmacopoeia. Ph. Eur. 2.6.14 — Bacterial Endotoxins. Método compendial para endotoxinas.
- European Pharmacopoeia. Ph. Eur. 5.1.10 — Guidelines for Using the Test for Bacterial Endotoxins. Recomendación de transición a rFC.
- ICH Harmonised Tripartite Guideline. Q3A(R2) — Impurities in New Drug Substances. International Council for Harmonisation.
- ICH Harmonised Tripartite Guideline. Q3B(R2) — Impurities in New Drug Products. International Council for Harmonisation.
- ICH Harmonised Tripartite Guideline. Q6B — Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products. International Council for Harmonisation.
- FDA Federal Register. Notice 2026-08552 — Bulks List for 503B Outsourcing Facilities: Semaglutide, Tirzepatide, Liraglutide. Department of Health and Human Services, mayo 2026.
- FDA. Drug Shortage Resolution: Tirzepatide (December 2024) and Semaglutide (February 2025). U.S. Food and Drug Administration.