Tu panel lipídico mide LDL-C. Tu riesgo cardiovascular real lo marca la ApoB. Y si nadie te ha medido la Lp(a), tienes un 20% de probabilidad de cargar con un factor genético que multiplica ese riesgo entre 2 y 4 veces, que no responde a estatinas y que no aparece en ningún análisis estándar.
El paradigma lipídico moderno ya no se ordena por la masa de colesterol que circula, sino por cuántas partículas aterogénicas penetran la pared arterial y se quedan retenidas. ApoB las cuenta todas. La Lp(a) añade una partícula con propiedades extra. El LDL-C aislado solo mide una parte, y en mucha gente miente.
En marzo de 2026, por primera vez, una guía de EE.UU. recomienda medir Lp(a) en todos los adultos y eleva la ApoB a objetivo de tratamiento. Esta es la pieza eje del cluster cardiovascular KRECE: qué mide cada métrica, en qué se diferencian, qué dice la causalidad y en qué orden actuar.
Una mujer de 48 años llega a una consulta de medicina preventiva. Delgada, hace deporte, come bien, no fuma. Su análisis muestra LDL-C de 105 mg/dL, triglicéridos de 90, HDL-C de 65. Su médico le dice que está «perfecta». Y sin embargo no sabe tres cosas que cambiarían su decisión clínica para los próximos 30 años: cuál es su ApoB (probablemente entre 95 y 110 mg/dL, por encima del percentil 60 poblacional), cuál es su Lp(a) (que nadie le ha medido y que tiene un 20% de probabilidad de estar alta), y cuál es su riesgo cardiovascular a lo largo de toda la vida, que es el que importa cuando se aspira a llegar lúcida a los 85.
Conviene separar tres preguntas que la práctica clínica funde: qué partícula causa el daño, con qué métrica se mide y en qué orden se interviene. Esta pieza es el cornerstone temático del cluster Prevención Cardiovascular de Precisión y desarrolla las dos métricas primarias del paradigma: ApoB y Lp(a). Es contenido editorial sobre un tema sensible (YMYL): toda escalada terapéutica corresponde a tu médico. Aquí está el criterio para entenderla.
-
N5 · consenso
ApoB cuenta partículas aterogénicas; el LDL-C solo mide colesterol. ApoB es mejor predictor que LDL-C, non-HDL-C y LDL-P, y está estandarizada desde 1994.
-
N5 · causal
La causalidad está cerrada: importa cuánto bajas la ApoB, no cómo la bajas. La exposición de por vida pesa mucho más que el fármaco tardío.
-
N5 · genético
La Lp(a) eleva el riesgo 2-4 veces y es genética. Las guías EAS/ESC 2022 y ACC/AHA 2026 piden medirla una vez en la vida en todos los adultos.
-
N3 · pipeline
Ya hay fármacos que bajan la Lp(a) un 80-99%, pero ningún ensayo de eventos ha leído todavía: Lp(a)HORIZON, el primero, tiene readout inminente.
VeredictoMide ApoB y Lp(a), corrige antes el motor metabólico, y escala la farmacología en orden. El panel que no incluye estas dos métricas está incompleto.
El riesgo no lo marca el colesterol que circula, sino cuántas partículas aterogénicas penetran la pared.
El daño arterial no lo causa la masa de colesterol que viaja por la sangre. Lo causan las partículas que cruzan el endotelio y quedan retenidas en la pared. Son dos variables distintas que se mueven de forma distinta en pacientes distintos, y ahí está toda la diferencia entre el paradigma de los años 80 y el de la década actual.
Qué mide el LDL-C y qué se le escapa
El colesterol LDL (LDL-C) es la masa de colesterol que contienen las partículas LDL del plasma. Es el «número del colesterol» que casi todo el mundo conoce, y el que la cardiología usó durante décadas. Pero el LDL-C habitual no se mide: se calcula con la fórmula de Friedewald a partir del colesterol total, el HDL-C y los triglicéridos. Esa estimación pierde precisión cuando los triglicéridos pasan de 400 mg/dL y cuando el LDL-C es muy bajo, los dos escenarios donde más importa acertar. Y, sobre todo, ignora cuántas partículas hay: dos personas con el mismo LDL-C pueden tener cargas de partículas muy diferentes.
Por qué la ApoB cuenta las partículas que importan
Cada lipoproteína aterogénica (quilomicrones residuales, VLDL, IDL, LDL y Lp(a)) lleva exactamente una molécula de apolipoproteína B. Es el esqueleto estructural de la partícula, y siempre hay una por partícula. Medir la concentración plasmática de ApoB equivale, por tanto, a contar directamente el número total de partículas aterogénicas circulantes [1]. El HDL no lleva ApoB, así que no entra en la cuenta. ApoB total = ApoB en quilomicrones + VLDL + IDL + LDL + Lp(a): toda la carga aterogénica en un solo número. El LDL-C es solo un subconjunto de esa carga, y un subconjunto cuyo tamaño depende del colesterol medio por partícula, una variable que el LDL-C aislado no captura.
ApoB vs Lp(a) vs LDL: qué mide exactamente cada uno
Las tres no son lo mismo y conviene fijarlo de una vez. El LDL-C mide masa de colesterol; la ApoB cuenta partículas; la Lp(a) es una partícula concreta con propiedades añadidas. Esta es la tabla que ordena las tres:
| Métrica | Qué mide | Medida o calculada | Evidencia |
|---|---|---|---|
| ApoB | Número total de partículas aterogénicas (todas) | Medida directa, estandarizada (IFCC 1994) | N5 superior |
| Lp(a) | Una partícula LDL + apo(a): aterogénica, proinflamatoria y protrombótica | Medida directa (mg/dL o nmol/L) | N5 causal |
| LDL-C | Masa de colesterol dentro de las partículas LDL | Calculada (Friedewald o Martin/Hopkins) | N5 proxy útil |
| No-HDL-C | Colesterol total menos HDL-C (proxy de carga total) | Calculada | N5 intermedio |
La jerarquía metodológica que establece el paradigma moderno es clara: ApoB > LDL-P > no-HDL-C > LDL-C. KRECE recomienda medir ApoB; donde no se pueda, no-HDL-C como segunda opción. El LDL-C aislado es la peor de las cuatro, aunque sea la que viene de serie en el análisis.
El cambio en una frase: el riesgo cardiovascular no se ordena por la masa de colesterol que circula, sino por el número de partículas aterogénicas que penetran y se retienen en la pared arterial. ApoB las cuenta. LDL-C no.
ApoB es mejor predictor que LDL-C, non-HDL-C y LDL-P: lo dice la evidencia, no la opinión.
Allan Sniderman (McGill) es la voz académica del cambio hacia ApoB como métrica primaria. Su revisión en JAMA Cardiology de 2019 sintetiza tres décadas de literatura comparada y concluye sin ambigüedad que la ApoB supera a LDL-C, no-HDL-C y al recuento de partículas LDL como predictor de riesgo [2]. No es opinión: es la síntesis de la evidencia.
Las tres razones técnicas
La superioridad de la ApoB tiene tres fundamentos concretos. Primero: cuenta partículas directamente, mientras que el LDL-C estima masa de colesterol. Segundo: está estandarizada internacionalmente desde 1994 con material de referencia certificado por la IFCC, mientras que el LDL-C habitual es calculado. Tercero: captura la carga aterogénica total, incluidos los remanentes de VLDL e IDL, que el LDL-C no incluye y que son clínicamente relevantes en quien tiene los triglicéridos altos. A esto se suma una ventaja práctica que las guías subrayan: la ApoB no se altera con el ayuno, así que no exige análisis en ayunas.
Cuándo falla Friedewald, y por qué Martin/Hopkins lo mejora
La fórmula de Friedewald (LDL-C = colesterol total – HDL-C – TG/5) pierde fiabilidad con triglicéridos por encima de 400 mg/dL y subestima el LDL-C cuando este es muy bajo, frecuente en tratamiento intensivo con estatinas o PCSK9. La guía ACC/AHA 2026 añade un matiz importante: cuando se usa el cálculo Martin/Hopkins en lugar de Friedewald, la discordancia entre LDL-C y ApoB se reduce de forma marcada [8]. Es decir, parte del problema del LDL-C es el método de cálculo, no solo el concepto. Aun así, al ser una medida directa y no calculada, la ApoB mantiene precisión donde cualquier fórmula tropieza.
El cociente ApoB/ApoA-I, ¿sirve para algo?
El cociente ApoB/ApoA-I enfrenta las partículas aterogénicas (ApoB) con las antiaterogénicas (ApoA-I, la proteína principal del HDL) en un solo número, y es un predictor de riesgo sólido en grandes cohortes como INTERHEART. Dicho esto, la posición de KRECE es preferir la ApoB absoluta como diana: el cociente mezcla numerador y denominador y diluye lo accionable, porque lo que se trata es bajar la ApoB, no «mejorar un ratio». El cociente sirve para estratificar riesgo; la ApoB sola sirve para fijar objetivos y seguir el tratamiento. Si tu informe trae el cociente, úsalo como semáforo, pero pide el valor absoluto de ApoB para decidir.
En qué orden medir todo esto
El paradigma no pide pedirlo todo a la vez. Pide medir lo que cambia decisiones. La ApoB sustituye al LDL-C como métrica de seguimiento; la Lp(a) se mide una vez; el resto del panel contextualiza. El orden completo, con qué medir y en qué secuencia, lo desarrollamos en la guía de biomarcadores de longevidad. Aquí basta con la regla: ApoB para seguir, Lp(a) una vez, y el motor metabólico antes que ninguno de los dos.
Puedes tener el LDL-C normal y el riesgo alto: eso es la discordancia.
La discordancia ApoB-LDL es la situación en la que ApoB y LDL-C colocan al mismo paciente en percentiles de riesgo distintos. El perfil típico: ApoB en rango alto y LDL-C aparentemente normal. No es excepcional, es frecuente, y el panel estándar no la detecta.
Qué es y a quién afecta
Las cifras dependen del método de cálculo del LDL-C, y conviene decirlo con precisión. En la población general, la discordancia significativa se ha estimado entre el 20% y el 30% de los pacientes [4], y sube mucho en subpoblaciones metabólicas: en resistencia a la insulina, síndrome metabólico o diabetes tipo 2 puede alcanzar tres de cada cuatro. El matiz honesto de 2026: con métodos de estimación mejores (Martin/Hopkins) la prevalencia baja respecto a Friedewald [8]. La discordancia es real e importa, pero su magnitud no es un número fijo: depende de cómo se haya calculado el LDL-C.
El paciente con LDL-C normal y ApoB alta
El caso paradigma es la resistencia a la insulina. El hígado produce más VLDL ricas en triglicéridos que se convierten en LDL pequeñas y densas: partículas numerosas pero depletadas de colesterol. El LDL-C aparece en rango porque cada partícula lleva poco colesterol, pero la ApoB está alta porque hay muchas partículas. El LDL-C tranquiliza al médico cuando debería alarmarle. Es el patrón de la dislipemia diabetogénica y la razón de que gente con perfil «normal» desarrolle aterosclerosis agresiva.
En discordancia, el riesgo sigue a la ApoB
Sniderman demostró con cohortes amplias que en todo caso de discordancia el riesgo cardiovascular real sigue a la ApoB, no al LDL-C [2]. Es la decisión clínica más consecuente del paradigma. Quien decide la intensidad del tratamiento mirando solo el LDL-C en un paciente discordante subestima el riesgo y subdosifica. Es exactamente el frame que ordena el cluster cardiovascular KRECE: la ApoB es diana modificable con causalidad demostrada; el LDL-C es un proxy útil que, en discordancia, infravalora el riesgo de forma sistemática.
Discordancia entre ApoB y LDL-C no es lo mismo que la disociación entre homocisteína elevada y eventos. Discordancia = dos métricas distintas de la misma diana causal dan señales diferentes. Disociación = un marcador asociativo que no es diana modificable. Son dos cosas distintas, las dos importantes para el frame «marcador frente a diana» de KRECE.
Que el LDL y la ApoB causan la enfermedad cardiovascular está demostrado, no en discusión.
El consenso de la European Atherosclerosis Society de 2017, liderado por Brian Ference, integró evidencia experimental, epidemiológica y de aleatorización mendeliana en una sentencia: las pruebas genéticas y clínicas establecen de forma inequívoca que el LDL causa enfermedad cardiovascular aterosclerótica [5]. Es la afirmación causal más clara de la cardiología moderna y la base sobre la que se construye toda la prevención de precisión.
La aleatorización mendeliana como marco
El argumento se apoya en la aleatorización mendeliana. Las variantes genéticas que reducen el LDL desde el nacimiento (en genes como HMGCR, NPC1L1, PCSK9 o LDLR) funcionan como instrumentos naturales que simulan ensayos de duración vital: son aleatorias en la población, independientes del estilo de vida y operan durante toda la vida. Producen conocimiento causal que ningún ensayo clínico puede generar, porque ningún ensayo dura 60 años ni arranca al nacer [5].
Exposición de por vida frente a fármaco tardío
La consecuencia operativa es directa. Bajar 1 mmol/L de LDL-C (unos 39 mg/dL) reduce el riesgo relativo de eventos en torno a un 25% a 5 años con fármacos. Pero el mismo descenso mantenido de por vida por variantes genéticas reduce el riesgo entre un 50% y un 80%. La diferencia es el efecto acumulado de 50 años de exposición frente a 5 de tratamiento tardío. La lógica de «intervenir pronto cuando se detecta» no es opinable: es estructural. Las guías ESC desplazaron los objetivos a ApoB <65 mg/dL en muy alto riesgo, <80 en alto y <100 en moderado; la ACC/AHA 2026 los alinea con sus metas de LDL en <55, <70 y <90 mg/dL según riesgo [8].
Ference 2019: 654.783 personas confirman el principio
En 2019, Ference publicó en JAMA el análisis que cerró el debate clínico. Agregó 63 cohortes y estudios caso-control entre 1948 y 2017: 654.783 participantes y 91.129 casos de cardiopatía coronaria [6]. Comparó variantes del gen LPL (que baja sobre todo triglicéridos) con variantes del gen LDLR (que baja sobre todo LDL-C). El resultado fue casi idéntico: por cada 10 mg/dL menos de ApoB, el odds ratio de cardiopatía fue 0,771 para LPL y 0,773 para LDLR. Tras ajustar por ApoB, la asociación de triglicéridos y de LDL-C con la enfermedad se desvaneció, y solo la ApoB conservó significancia. Traducción clínica: importa cuánto bajas la ApoB, no cómo la bajas. La métrica de seguimiento debe ser ApoB.
La Lp(a) eleva el riesgo 2-4 veces, es genética y casi nadie la mide.
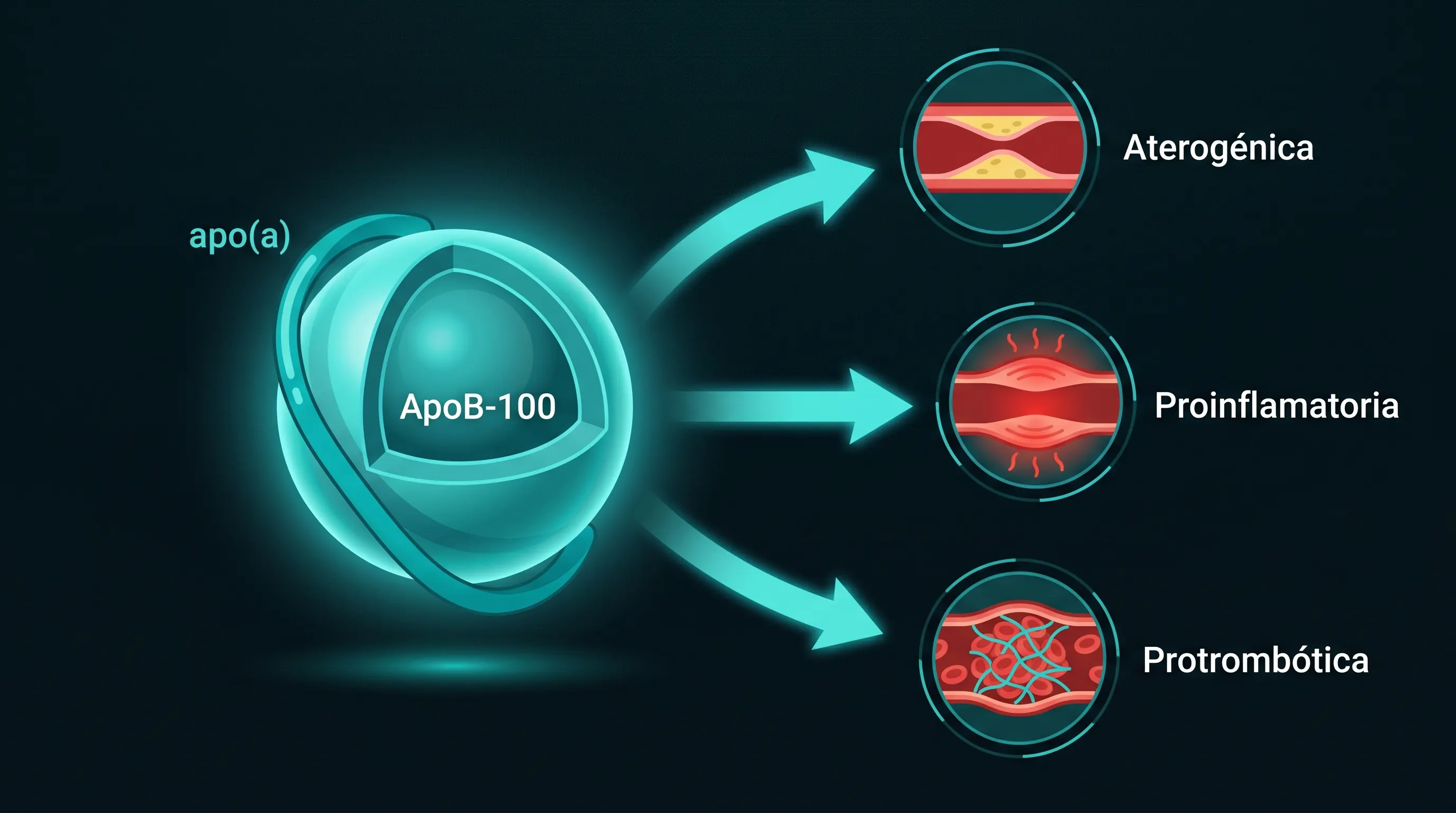
La lipoproteína(a) es, estructuralmente, una partícula LDL con una apolipoproteína(a) unida de forma covalente a la ApoB-100 [7]. Esa apo(a) comparte similitud estructural con el plasminógeno, y de ahí salen sus consecuencias. La Lp(a) conserva todo lo aterogénico del LDL y añade tres propiedades que la hacen peligrosa de una forma que ninguna otra partícula reúne.
Por qué es excepcionalmente peligrosa
La Lp(a) es a la vez aterogénica (como toda partícula con ApoB, penetra el endotelio y forma placa), proinflamatoria (transporta fosfolípidos oxidados que generan placas más vulnerables a la rotura) y protrombótica (la apo(a) compite con el plasminógeno por la fibrina, inhibe la fibrinólisis endógena y favorece la persistencia de microtrombos) [7]. Esa tercera vía conecta directamente con el eje fibrinolítico que desarrolla la pieza sobre natoquinasa del cluster.
Es genética y estable de por vida
Los niveles plasmáticos de Lp(a) están determinados genéticamente en más de un 90% por el gen LPA (cromosoma 6) [9]. El número variable de repeticiones del kringle IV-2 fija el tamaño de la apo(a) y, de forma inversa, el nivel de Lp(a): isoformas pequeñas, niveles altos. La consecuencia práctica es contundente: la Lp(a) es estable a lo largo de la vida (fluctúa poco con inflamación crónica o menopausia) y no responde a dieta, ejercicio, pérdida de peso ni estatinas. Solo la mueven fármacos específicos, que veremos en la sección 06.
Medir una vez en la vida: lo que dicen EAS/ESC 2022 y ACC/AHA 2026
El consenso EAS/ESC de 2022, liderado por Florian Kronenberg, recomienda medir la Lp(a) al menos una vez en la vida en todos los adultos: no es cribado selectivo, es universal [10]. La justificación es que el nivel es genéticamente estable, la prueba es barata y conocer el valor cambia el manejo del resto de factores. La novedad de 2026 cierra el círculo: la guía ACC/AHA, por primera vez en EE.UU., recomienda la medición universal de Lp(a) una vez en todos los adultos con designación Clase I, y añade el cribado en cascada de familiares de primer grado cuando la Lp(a) está alta [8]. El consenso ya no es solo europeo. Lo que sigue faltando no es la recomendación, sino su aplicación: en la consulta europea e hispana la Lp(a) se sigue sin pedir. Para el detalle de mecanismo y pipeline, el cornerstone temático es Lipoproteína(a): factor de riesgo cardiovascular, evidencia 2026.
Qué nivel deberías tener
El consenso EAS/ESC 2022 define umbrales operativos claros. Aproximadamente el 20% de la población mundial tiene Lp(a) por encima de 50 mg/dL (uno de cada cinco adultos), casi siempre sin saberlo, porque no entra en el panel estándar de ningún país:
| Nivel Lp(a) | Equivalencia molar | Estratificación clínica |
|---|---|---|
| <30 mg/dL | <75 nmol/L | Riesgo bajo. Sin acción específica. |
| 30-50 mg/dL | 75-125 nmol/L | Riesgo elevado. Intensificar los demás factores. |
| >50 mg/dL | >125 nmol/L | Significativo (20% población). Manejo agresivo. |
| >90 mg/dL | >200 nmol/L | Riesgo severo. Candidato prioritario a ASO/siRNA cuando se aprueben. |
Estenosis aórtica y tromboembolismo venoso
La Lp(a) elevada no solo causa aterosclerosis arterial. Promueve la calcificación de la válvula aórtica estimulando proteínas osteogénicas en las células intersticiales valvulares, que se transforman en células formadoras de hueso: el hazard ratio para estenosis aórtica se sitúa entre 2 y 4 [11]. Es el factor de riesgo no modificable más sólidamente asociado a la estenosis aórtica calcificada degenerativa, la causa más frecuente de recambio valvular en adultos. Y por la vía protrombótica, la apo(a) eleva también el riesgo de tromboembolismo venoso (HR en torno a 3) [12], una dimensión clínicamente subestimada.
Qué baja la ApoB, qué baja la Lp(a) y dónde está la frontera.
Cada fármaco hace una cosa distinta, y conviene saber qué modifica cada uno, sobre todo qué no modifica. La regla de fondo, ya demostrada, es que el beneficio es proporcional al descenso absoluto de ApoB, sea cual sea la vía.
Estatinas, ezetimiba y PCSK9
Las estatinas reducen el LDL-C un 30-50% y arrastran la ApoB, pero no bajan la Lp(a) (incluso pueden subirla algo) [7]; siguen siendo la base del tratamiento, con reposición de CoQ10 por la depleción vía mevalonato. La ezetimiba añade reducción sobre la estatina y demostró menos eventos en IMPROVE-IT [14]. Los inhibidores de PCSK9 (evolocumab, alirocumab, inclisiran) bajan el LDL-C un 50-70% adicional y la Lp(a) un 20-30%, el único efecto sobre Lp(a) disponible hoy fuera del pipeline; FOURIER lo validó con outcomes duros [13]. El detalle está en la pieza sobre PCSK9.
El pipeline contra la Lp(a): el campo más dinámico de 2026
La primera generación de fármacos específicos contra la Lp(a) está en fases avanzadas. Reducen el biomarcador de forma espectacular, pero ningún ensayo de eventos ha leído todavía: esa es la frontera exacta.
| Fármaco | Clase · vía | Reducción Lp(a) | Fase · ensayo · sponsor |
|---|---|---|---|
| Pelacarsen | ASO · subcutáneo mensual | ~80% | Fase 3 Lp(a)HORIZON (n=8.323, prevención secundaria) · Novartis/Ionis · readout inminente |
| Olpasiran | siRNA · subcutáneo trimestral | hasta ~99% | Fase 3 OCEAN(a) · Amgen |
| Lepodisiran | siRNA · subcutáneo semestral | ~94% | Fase 3 ALPACA · Eli Lilly |
| Muvalaplin | Molécula pequeña oral diaria | ~86% | Fase 2/3 · Eli Lilly · impide el ensamblaje de la Lp(a) |
Lp(a)HORIZON con pelacarsen es el ensayo que decidirá si bajar la Lp(a) reduce eventos en humanos. Son 8.323 pacientes en prevención secundaria con Lp(a) basal ≥70 mg/dL, asignados a pelacarsen mensual o placebo, con MACE como objetivo. El ensayo completó seguimiento en febrero de 2026 y su topline se espera en la primera mitad del año: a fecha de esta revisión aún no se ha publicado. Si demuestra reducción de eventos proporcional al descenso del biomarcador, por primera vez en seis décadas de conocer la Lp(a) tendremos un fármaco aprobado para bajarla. Ese readout es la próxima revisión editorial obligatoria de esta pieza.
Lo que no funciona: la niacina
La niacina baja la Lp(a) un 20-30% y los triglicéridos, pero dos ensayos grandes, AIM-HIGH y HPS2-THRIVE, no demostraron reducción de eventos al añadirla a estatinas, con efectos adversos relevantes [15]. KRECE no la recomienda como intervención de primera línea contra la Lp(a): el mecanismo es real, la evidencia de outcomes es nula.
La resistencia a la insulina fabrica las partículas: corrige el motor antes que la farmacología.
La resistencia a la insulina no es un factor de riesgo más sumado al perfil lipídico: es el motor que lo genera. Y opera por dos frentes a la vez, fabricando partículas y preparando la pared donde se depositan.
Cómo la hiperinsulinemia produce partículas
La hiperinsulinemia compensatoria estimula al hígado a producir más VLDL ricas en triglicéridos, que se convierten en IDL y finalmente en LDL pequeñas y densas a través de la cascada lipolítica [3]. Más VLDL hepática significa más partículas ApoB circulantes y más riesgo. Esas partículas pequeñas penetran el endotelio con más facilidad y quedan retenidas mejor: cada una lleva menos colesterol, pero el daño por partícula es mayor. Es el origen mecánico de la discordancia de la sección 03.
Y prepara la pared: endotelio y glicación
La hiperinsulinemia crónica produce estrés oxidativo en el endotelio: reduce el óxido nítrico (vasodilatador y antiagregante), eleva citoquinas proinflamatorias que reclutan monocitos a la pared, y aumenta la endotelina-1 vasoconstrictora. Para el eje inflamatorio crónico, ver la pieza sobre inflammaging. Además, en hiperglucemia, la ApoB sufre glicación no enzimática y se adhiere mejor a la matriz subendotelial: más glicación, más retención, más placa. Por eso quien tiene LDL-C «normal» pero resistencia a la insulina desarrolla aterosclerosis agresiva; sus partículas no son más, pero se pegan mejor.
El motor antes que la farmacología: cerrar la fábrica de partículas (corregir la resistencia a la insulina) es metabólicamente más eficiente que barrer el producto con fármacos. Ejercicio de fuerza y resistencia, reducción de grasa visceral, control de carbohidratos refinados. Reevaluar la ApoB a los 3-6 meses y modular la intensidad farmacológica según el resultado.
El orden correcto: motor, métricas, estatinas, PCSK9, anti-Lp(a) y adyuvantes.
La cadena causal se traduce en una secuencia. No es una lista de opciones a la carta: es un orden, y el orden importa.
Pasos 1 y 2: el motor y luego las métricas
Paso 1, corregir la resistencia a la insulina como base. Métricas operativas: HOMA-IR <1,5, HbA1c <5,4%, cociente TG/HDL <1,5, ausencia de esteatosis metabólica. Intervención no farmacológica primero, reevaluar a los 3-6 meses. Paso 2, medir ApoB y Lp(a) tras esa optimización. Objetivos de ApoB <90 / <80 / <65 mg/dL según riesgo (ESC), alineados por ACC/AHA 2026 con sus metas de LDL en <90 / <70 / <55 mg/dL [8]. La Lp(a), una sola vez: si >50 mg/dL, intensificar el resto; si >90, candidato prioritario a anti-Lp(a).
Pasos 3 a 5: la escalada farmacológica
Paso 3, estatina si la ApoB persiste alta tras optimizar el estilo de vida, con reposición de CoQ10; añadir ezetimiba si el LDL-C es muy alto o hay enfermedad establecida. Paso 4, inhibidor de PCSK9 si la ApoB sigue por encima de objetivo en alto riesgo tras estatina máxima tolerada más ezetimiba; razonable también con Lp(a) >90 mg/dL aún con LDL-C en rango, por su efecto parcial sobre la Lp(a). Paso 5, ASO/siRNA contra la Lp(a) para Lp(a) genéticamente elevada con riesgo persistente, cuando se aprueben; hasta el readout de Lp(a)HORIZON, la hipótesis se apoya en la mecánica y la aleatorización mendeliana, no en outcomes.
Paso 6: adyuvantes según el eje del riesgo residual
Sobre ese esqueleto, dos adyuvantes con evidencia humana se posicionan de forma transversal. Natoquinasa para el eje fibrinolítico, que desarrolla la pieza dedicada, y EPA/DHA marinos en dosis terapéutica para el eje inflamatorio residual, en la pieza sobre omega-3. No sustituyen a estatinas ni a PCSK9: los complementan en ejes distintos. Como contraste editorial, conviene recordar qué cosas son marcador y no diana, como la testosterona baja y la mortalidad: asociación sí, diana modificable por demostrar.
Preguntas frecuentes
¿Qué diferencia hay entre la ApoB y la Lp(a)?
La ApoB es una medida del número total de partículas aterogénicas (cada partícula lleva una molécula de ApoB), mientras que la Lp(a) es una partícula concreta: una LDL con una apolipoproteína(a) añadida. Dicho de otro modo, la Lp(a) es una de las partículas que la ApoB cuenta, pero con propiedades extra (proinflamatoria y protrombótica) y un origen casi totalmente genético. Conviene medir las dos: la ApoB para la carga global, la Lp(a) para el riesgo genético independiente.
¿Es mejor medir la ApoB o el colesterol LDL?
La ApoB es mejor predictor de riesgo que el LDL-C: cuenta partículas en lugar de estimar masa de colesterol, está estandarizada internacionalmente y no se altera con el ayuno. El LDL-C habitual es calculado y, en personas con triglicéridos altos o resistencia a la insulina, puede aparecer normal mientras la ApoB está alta. Donde se pueda medir ApoB, es la métrica de elección para decidir y seguir el tratamiento; el LDL-C aislado es la peor de las opciones disponibles.
¿Qué es el cociente ApoB/ApoA-I y para qué sirve?
Es la relación entre las partículas aterogénicas (ApoB) y las antiaterogénicas (ApoA-I, la proteína principal del HDL), y es un predictor de riesgo sólido en grandes estudios. Sirve bien para estratificar el riesgo, pero KRECE prefiere usar la ApoB absoluta como diana: lo que se trata es bajar la ApoB, no mejorar un ratio que mezcla numerador y denominador. Si tu informe trae el cociente, úsalo como semáforo y pide el valor absoluto de ApoB para decidir.
¿Cuántas veces hay que medir la Lp(a)?
Una sola vez en la vida basta para la mayoría de las personas. Como el nivel está determinado en más del 90% por la genética y se mantiene estable, no hace falta repetir la prueba. Tanto el consenso EAS/ESC de 2022 como la guía ACC/AHA de 2026 recomiendan medirla al menos una vez en todos los adultos, y hacer cribado en cascada de los familiares de primer grado si el valor está elevado.
¿Se puede bajar la Lp(a)?
Con dieta, ejercicio o estatinas, no: la Lp(a) no responde a esas intervenciones. Los inhibidores de PCSK9 la bajan un 20-30%, un efecto modesto. La gran novedad es el pipeline específico (pelacarsen, olpasiran, lepodisiran, muvalaplin), que la reduce entre un 80% y un 99%. Eso sí: a fecha de hoy ningún ensayo ha demostrado todavía que bajar la Lp(a) reduzca eventos cardiovasculares; el primero en leer resultados será Lp(a)HORIZON.
¿Qué nivel de ApoB es el objetivo?
Depende del riesgo. Las guías europeas fijan ApoB <65 mg/dL en muy alto riesgo, <80 en alto y <100 en moderado; la guía ACC/AHA de 2026 alinea la ApoB con sus metas de LDL en <55, <70 y <90 mg/dL según la categoría. La relación entre ApoB y riesgo no tiene un umbral inferior conocido: cada reducción proporcional baja el riesgo de forma proporcional. La cifra exacta la fija tu médico según tu contexto clínico.
- Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116(16):1832-1844. PubMed. La retención subendotelial de partículas con ApoB es el evento iniciador de la aterosclerosis.
- Sniderman AD, Thanassoulis G, Glavinovic T, Navar AM, Pencina M, Catapano A, Ference BA. Apolipoprotein B Particles and Cardiovascular Disease: A Narrative Review. JAMA Cardiol. 2019;4(12):1287-1295. PubMed. Referencia metodológica del paradigma ApoB: superioridad sobre LDL-C, no-HDL-C y LDL-P.
- Krauss RM. Lipoprotein subfractions and cardiovascular disease risk. Curr Opin Lipidol. 2010;21(4):305-311. PubMed. Partículas LDL pequeñas y densas, resistencia a la insulina y riesgo por partícula.
- Sniderman AD, St-Pierre AC, Cantin B, Dagenais GR, Després JP, Lamarche B. Concordance/discordance between plasma apolipoprotein B levels and the cholesterol indexes of atherosclerotic risk. Am J Cardiol. 2003;91(10):1173-1177. PubMed. Cuantificación de la discordancia ApoB-LDL.
- Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38(32):2459-2472. PubMed. La sentencia causal del consenso EAS.
- Ference BA, Kastelein JJP, Ray KK, et al. Association of Triglyceride-Lowering LPL Variants and LDL-C-Lowering LDLR Variants With Risk of Coronary Heart Disease. JAMA. 2019;321(4):364-373. PubMed. n=654.783, 91.129 casos, 63 cohortes 1948-2017: el beneficio es proporcional al cambio en ApoB.
- Tsimikas S. A Test in Context: Lipoprotein(a): Diagnosis, Prognosis, Controversies, and Emerging Therapies. J Am Coll Cardiol. 2017;69(6):692-711. PubMed. Estructura, mecanismos aterogénico, proinflamatorio y protrombótico de la Lp(a), y por qué las estatinas no la bajan.
- Blumenthal RS, Morris PB, Gaudino M, et al. 2026 ACC/AHA/Multisociety Guideline on the Management of Dyslipidemia: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. Publicado online 13 marzo 2026. doi:10.1016/j.jacc.2025.11.016. JACC. Medición universal de Lp(a) (Clase I), objetivos de ApoB alineados con LDL (<55/70/90 mg/dL), Martin/Hopkins y ecuaciones PREVENT-ASCVD.
- Boerwinkle E, Leffert CC, Lin J, Lackner C, Chiesa G, Hobbs HH. Apolipoprotein(a) gene accounts for greater than 90% of the variation in plasma lipoprotein(a) concentrations. J Clin Invest. 1992;90(1):52-60. PubMed. Determinación genética de la Lp(a) por el gen LPA.
- Kronenberg F, Mora S, Stroes ESG, et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: a European Atherosclerosis Society consensus statement. Eur Heart J. 2022;43(39):3925-3946. PubMed. Consenso EAS/ESC 2022: medir Lp(a) una vez en la vida; umbrales operativos.
- Kamstrup PR, Tybjærg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014;63(5):470-477. PubMed. Asociación de Lp(a) elevada con estenosis aórtica calcificada.
- Dentali F, Gessi V, Marcucci R, Gianni M, Grandi AM, Franchini M. Lipoprotein(a) as a Risk Factor for Venous Thromboembolism: A Systematic Review and Meta-analysis. Semin Thromb Hemost. 2017;43(6):614-620. PubMed. Lp(a) elevada y riesgo de tromboembolismo venoso.
- Sabatine MS, Giugliano RP, Keech AC, et al; FOURIER Steering Committee and Investigators. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med. 2017;376(18):1713-1722. PubMed. PCSK9: reducción de LDL-C y de eventos con outcomes duros.
- Cannon CP, Blazing MA, Giugliano RP, et al; IMPROVE-IT Investigators. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N Engl J Med. 2015;372(25):2387-2397. PubMed. Más reducción de LDL-C produce menos eventos, incluso a niveles bajos.
- HPS2-THRIVE Collaborative Group; Landray MJ, Haynes R, Hopewell JC, et al. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371(3):203-212. PubMed. La niacina no reduce eventos al añadirla a estatinas, con efectos adversos relevantes.