La prevención cardiovascular de 2026 ya no se entiende como «colesterol alto o bajo». Se entiende como cuántas partículas aterogénicas hay (ApoB), qué genética las multiplica (Lp(a)), qué metabolismo subyace, y qué intervenciones mueven eventos reales frente a las que solo mueven un número.
El error cardiovascular más repetido de los últimos cuarenta años ha sido confundir un marcador asociado con el riesgo con una diana que, modificada, reduce el riesgo. La homocisteína, el HDL-C y la testosterona son los ejemplos de manual. Distinguir una cosa de la otra es la diferencia entre la prevención poblacional de los noventa y la de precisión.
En marzo de 2026, la guía ACC/AHA de EE.UU. convergió por fin con Europa: Lp(a) universal Clase I, objetivos de ApoB, ecuaciones de riesgo nuevas. Este es el editorial ancla del cluster cardiovascular de KRECE: el filtro que separa marcadores de dianas, y las siete piezas que lo construyen.
Un hombre de 52 años llega a una consulta de medicina preventiva. Lleva tres años con una estatina de intensidad moderada. Su LDL-C está en 70 mg/dL, su HDL en 48, sus triglicéridos en 165. Su médico le dice que está bien y su calculadora de riesgo a 10 años lo sitúa en riesgo bajo. Y sin embargo sigue sin saber tres cosas que cambiarían la decisión: cuál es su ApoB (probablemente 110-120 mg/dL, en discordancia con su LDL-C), cuál es su Lp(a) (que nadie le ha medido), y cuál es su riesgo a 30 años, que es el que importa cuando uno tiene 52 y aspira a llegar lúcido a los 85.
Este editorial parte de ese paciente y desemboca en el marco que ordena toda la línea cardiovascular de KRECE. Es la pieza ancla del cluster Prevención cardiovascular de precisión y conecta siete satélites: ApoB y Lp(a), el cornerstone de Lipoproteína(a), PCSK9, estatinas y CoQ10, natoquinasa, homocisteína y testosterona y mortalidad. Todos bajo el mismo principio: marcador asociativo robusto no es lo mismo que diana modificable. Es contenido editorial sobre un tema sensible (YMYL); toda intervención corresponde a tu médico.
-
N5 · causal
El filtro KRECE: solo es diana modificable lo que la aleatorización mendeliana y los ensayos confirman como causa. LDL-C, ApoB, Lp(a) y PCSK9 lo cumplen. El resto son marcadores.
-
N5 · métrica
La ApoB es la métrica primaria y la Lp(a) se mide una vez en la vida. Desde 2026 lo respaldan tanto las guías europeas como las de EE.UU.
-
N4 · residual
Con el LDL en rango persiste hasta el 70% del riesgo: se reparte en cuatro componentes residuales (aterogénico, inflamatorio, trombótico y metabólico), cada uno con su estrategia.
-
N5 · marcador
HDL-C, homocisteína y testosterona no son dianas: asocian con riesgo pero modificarlos no reduce eventos. Confundirlos cuesta tiempo, dinero y criterio.
VeredictoDistingue marcador de diana, mide ApoB y Lp(a), corrige antes el motor metabólico, e interviene solo donde hay evidencia de eventos.
La prevención cardiovascular de 2026 ya no se ordena por el colesterol, sino por partículas, genética y causalidad.
El modelo que la mayoría de médicos aprendió en la facultad se construyó sobre Framingham: un calculador te daba una probabilidad de evento a 10 años, aplicabas estatinas si superaba un umbral, y el LDL-C era la diana. Fue eficaz a escala poblacional y la mortalidad cardiovascular cayó durante décadas. Pero arrastra tres límites que en 2026 son insostenibles.
Qué era la prevención cardiovascular del siglo XX
Trabajaba con promedios poblacionales aplicados a individuos, usaba un horizonte de 10 años incompatible con la longevidad, y se apoyaba en un biomarcador indirecto (LDL-C, masa de colesterol) en vez de en lo que de verdad daña la arteria, que es el número de partículas que la penetran. Servía para decidir en poblaciones; falla cuando el paciente concreto se aparta del promedio, que es casi siempre.
Qué es la prevención cardiovascular de precisión
Se define por tres ejes. Estratificación individual con biomarcadores de causalidad genética validada (ApoB, Lp(a)) e imagen subclínica (calcio coronario), no solo un calculador estadístico. Intervención de mecanismo causal demostrado: se modifica solo lo que la aleatorización mendeliana o los ensayos han demostrado que mueve eventos, no biomarcadores intermedios. Y horizonte temporal extendido al riesgo a 30-40 años. Un varón de 40 años con ApoB alta puede tener riesgo a 10 años por debajo del 5% y riesgo a lo largo de la vida superior al 50%, y eso cambia por completo la decisión de tratar o esperar.
Por qué el LDL-C ya no basta como métrica primaria
El LDL-C mide la masa de colesterol que circula en las partículas LDL. Pero el daño no lo causa la masa, lo causa el número de partículas que penetran y se retienen. Dos pacientes con el mismo LDL-C de 100 mg/dL pueden tener cargas de partículas radicalmente distintas: el de partículas pequeñas y densas (típico de la resistencia a la insulina) tiene muchas más partículas y mucho más riesgo, y el LDL-C no lo ve. Además, el LDL-C habitual es calculado y pierde precisión con triglicéridos altos o LDL muy bajo.[1] Por eso la métrica primaria moderna es la ApoB, que se desarrolla en la sección 03.
El criterio que ordena todo: un marcador asociado al riesgo no es lo mismo que una diana que, modificada, lo reduce.
Este es el corazón editorial del cluster. La epidemiología observacional asocia. Los ensayos clínicos prueban que una intervención hace algo. La aleatorización mendeliana prueba que la exposición de por vida causa la enfermedad. Son tres niveles distintos de conocimiento y exigen una jerarquía distinta. Confundirlos es el error que ha costado cuarenta años de farmacología fallida.
Asociación robusta no es causalidad demostrada
El patrón del error se repite: una cohorte observa que un biomarcador asocia con eventos, se diseñan fármacos que lo modifican, los ensayos demuestran que no reducen eventos, y aun así se sigue tratando el biomarcador. La homocisteína y el HDL-C son los dos casos de manual del último decenio, y se desarrollan en la sección 06. Sir Austin Bradford Hill propuso en 1965 nueve criterios para juzgar si una asociación refleja causalidad (fuerza, consistencia, temporalidad, gradiente dosis-respuesta, plausibilidad, evidencia experimental, entre otros).[2] La ApoB los cumple casi todos; la homocisteína falla los decisivos.
Qué es la aleatorización mendeliana
La aleatorización mendeliana es un método que usa variantes genéticas asignadas al azar en la concepción como un ensayo natural de duración vital. Como esas variantes se reparten al azar, son independientes del estilo de vida y actúan desde el nacimiento, permiten estimar si una exposición (por ejemplo, un LDL-C alto de por vida) causa la enfermedad, evitando la confusión y la causalidad inversa que limitan a los estudios observacionales. En la práctica, si una variante que baja el LDL-C desde el nacimiento reduce los eventos en proporción a la magnitud y la duración del descenso, la causalidad queda demostrada. Es la herramienta que cerró el debate causal del LDL-C (Ference 2017) y de la Lp(a) (consenso EAS 2022), y la que descartó el HDL-C, la homocisteína y la testosterona endógena como dianas modificables.
La regla operativa: sin causalidad demostrada, no es diana
La regla que aplicamos en todo el cluster es simple: solo posicionamos como diana modificable lo que converge en observacional, aleatorización mendeliana y ensayo clínico. El resto son marcadores útiles para estratificar riesgo, no para guiar farmacología específica. La regla produce dos categorías limpias: dianas con outcomes demostrados (LDL-C, ApoB, Lp(a) por inhibición con ASO/siRNA, PCSK9) y marcadores que no son dianas (HDL-C, homocisteína, testosterona, ácido úrico). La diferencia es la que hay entre prescribir algo útil y prescribir algo caro.
ApoB y Lp(a): la causalidad genética como prueba definitiva.
Cada partícula aterogénica que circula (VLDL, IDL, LDL y Lp(a)) lleva exactamente una molécula de apolipoproteína B-100. La concentración de ApoB es, por tanto, un recuento directo del número de partículas capaces de infiltrar el endotelio. El LDL-C mide la masa de colesterol dentro de esas partículas, que depende tanto del número como del tamaño. Esa diferencia conceptual es la base del cambio de paradigma.
De la masa de colesterol al recuento de partículas
Allan Sniderman sintetizó en JAMA Cardiology (2019) por qué la ApoB supera a LDL-C, no-HDL-C y LDL-P: cuenta partículas directamente, está estandarizada desde 1994, no depende del ayuno y captura los remanentes que el LDL-C ignora.[1] El detalle clínico, con la comparación entre las tres métricas y el cociente ApoB/ApoA-I, está en la pieza dedicada a ApoB y Lp(a).
Discordancia ApoB-LDL: el riesgo silente
El concepto clínico más consecuente del paradigma es la discordancia: ApoB y LDL-C sitúan al paciente en percentiles de riesgo distintos. Afecta al 20-30% de la población general y hasta al 75% en síndrome metabólico, donde las partículas LDL están deplecionadas de colesterol (pequeñas y densas) y el LDL-C aparece en rango mientras la ApoB está alta. En todo caso de discordancia, el riesgo sigue a la ApoB, no al LDL-C. El LDL-C tranquiliza cuando debería alarmar.
Lp(a): el factor de riesgo genético independiente
La Lipoproteína(a) es una partícula LDL con una apo(a) añadida que le suma propiedades aterogénicas, proinflamatorias y protrombóticas. Sus niveles son genéticos en ~90% y no responden al estilo de vida. El consenso EAS/ESC 2022 recomienda medir Lp(a) una vez en la vida en todos los adultos,[4] y desde 2026 la guía ACC/AHA hace lo propio con designación Clase I.[5] Umbrales: >50 mg/dL riesgo relevante (un 20% de la población), >90 mg/dL severo. La profundidad está en el cornerstone de Lp(a).
Ference EAS 2017 y la exposición acumulada
El consenso EAS 2017, liderado por Brian Ference, integró estudios genéticos, epidemiológicos y de intervención en una sentencia: la evidencia establece de forma inequívoca que el LDL causa enfermedad cardiovascular.[3] El concepto operativo es la exposición acumulada: el riesgo es función del producto entre concentración de ApoB y años de exposición. Por eso un paciente de 35 años con ApoB de 120 mg/dL tiene más riesgo de por vida que uno de 65 con ApoB de 130: acumula tres décadas más. La lógica de intervenir pronto es estructural, no opinable.
El producto que importa es ApoB por años. Bajar 1 mmol/L de LDL-C reduce el riesgo un 25% a 5 años con fármacos, pero el mismo descenso mantenido de por vida por genética lo reduce entre un 50% y un 80%. La diferencia es 50 años de exposición frente a 5 de tratamiento tardío.
Con el LDL en rango persiste hasta el 70% del riesgo: cuatro componentes, cuatro estrategias.
El riesgo residual es la probabilidad de evento que persiste pese al tratamiento óptimo con estatinas y el LDL-C en rango. La reducción cardiovascular total con hipolipemiantes agresivos rara vez supera el 30%, así que el grueso del riesgo no se elimina bajando el LDL. Se reparte en cuatro componentes diferenciables, y cada uno exige su métrica y su intervención.
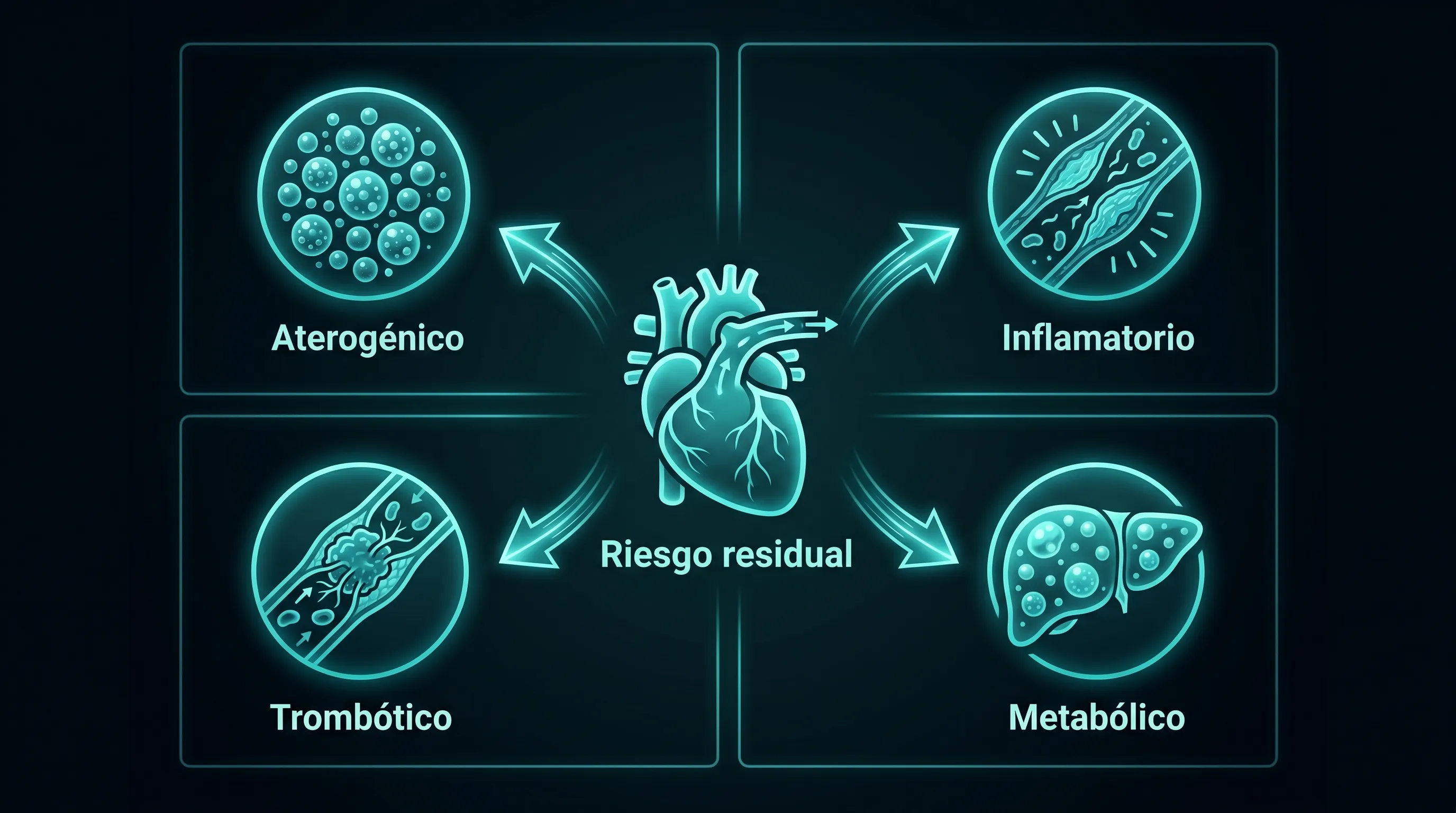
Residual aterogénico: remanentes, Lp(a) y triglicéridos
Cuando el LDL-C está controlado pero la ApoB sigue alta, la diferencia son partículas no-LDL que el LDL-C no cuenta: remanentes de VLDL, IDL y Lp(a). El colesterol remanente y los triglicéridos elevados añaden riesgo incluso en pacientes tratados,[6] y la Lp(a) multiplica el riesgo y no responde a estatinas. La diana es la ApoB por debajo de 60-80 mg/dL, no el LDL-C aislado.
Residual inflamatorio: CANTOS y la hsCRP
El estudio CANTOS (2017, n=10.061) demostró que canakinumab, un anticuerpo contra la IL-1β, reducía eventos recurrentes sin tocar el LDL-C: la primera confirmación causal de la vía inflamatoria.[7] El biomarcador operativo es la proteína C reactiva ultrasensible (objetivo <1 mg/L). La conexión es directa con la pieza sobre inflammaging, y la intervención con datos humanos más sólida en este eje son los EPA/DHA marinos.
Residual trombótico: la fibrinólisis ignorada
El tercer componente es la capacidad de disolver microtrombos sobre placas erosionadas. Marcadores: fibrinógeno, dímero D, ratio PAI-1/tPA. Es el componente más ignorado por la práctica estándar y el que justifica el interés de KRECE en agentes profibrinolíticos con datos humanos, en la pieza sobre natoquinasa. En la era post-COVID, el dímero D crónicamente elevado ha resultado un predictor independiente de eventos arteriales, lo que recoloca este eje en el centro.
Residual metabólico: hiperinsulinemia y MASLD
El cuarto componente es la base que produce las partículas y daña la pared. Marcadores: HOMA-IR (patológico >2,5), HbA1c (el riesgo sube linealmente desde 5,2%, antes del umbral diabético) y esteatosis hepática metabólica (MASLD). Es el motor del problema y se desarrolla en la sección 08.
Lo que sí mueve eventos cardiovasculares en ensayos rigurosos.
El filtro aplicado a la farmacología deja un puñado de intervenciones con outcomes demostrados. La regla de fondo: el beneficio es proporcional al descenso absoluto de ApoB, sea cual sea la vía.
Estatinas: el suelo, no el techo
Son el fármaco preventivo más probado de la historia: cada 1 mmol/L de descenso de LDL-C reduce un 20-22% los eventos vasculares mayores. Son la base. Lo que no hacen es bajar la Lp(a), cubrir el residual inflamatorio o trombótico, ni corregir la base metabólica. En muchos pacientes la estatina es el suelo del tratamiento, no el techo, y conviene reponer la CoQ10 que deplecionan, en la pieza sobre estatinas y CoQ10.
Inhibidores de PCSK9: el avance subutilizado
Evolocumab, alirocumab e inclisiran bajan el LDL-C un 50-60% adicional sobre estatina y reducen la Lp(a) un 25-30%. FOURIER demostró reducción de eventos con outcomes duros,[8] y la validación genética viene de las variantes naturales de pérdida de función de PCSK9, que confieren protección coronaria de por vida.[9] La prescripción real en España, Europa y LATAM es una fracción de los candidatos elegibles. Detalle en la pieza sobre PCSK9.
Natoquinasa: la dimensión fibrinolítica con N4
La natoquinasa es una serina proteasa del nattô con evidencia clínica N4 en múltiples outcomes cardiovasculares simultáneos (LDL, placa carotídea, presión arterial). KRECE la posiciona como adyuvante de primera línea en optimización preventiva, especialmente en el contexto post-viral con dímero D elevado. Los datos y dosis están en la pieza dedicada.
El pipeline contra la Lp(a)
El campo más dinámico de 2026 es la primera generación de fármacos específicos contra la Lp(a). Reducen el biomarcador de forma espectacular, pero ningún ensayo de eventos ha leído todavía:
| Fármaco | Clase · vía | Reducción Lp(a) | Fase · ensayo · sponsor |
|---|---|---|---|
| Pelacarsen | ASO · subcutáneo mensual | ~80% | Fase 3 Lp(a)HORIZON (n=8.323) · Novartis/Ionis · readout inminente |
| Olpasiran | siRNA · trimestral | hasta ~99% | Fase 3 OCEAN(a) · Amgen |
| Lepodisiran | siRNA · semestral | ~94% | Fase 3 ALPACA · Eli Lilly |
| Muvalaplin | Oral diario | ~86% | Fase 2/3 · Eli Lilly · impide el ensamblaje |
Lp(a)HORIZON con pelacarsen es el ensayo que decidirá si bajar la Lp(a) reduce eventos en humanos.[10] Cerró seguimiento en febrero de 2026 y su topline se espera en la primera mitad del año: a fecha de esta revisión aún no se ha publicado. Si demuestra reducción de eventos proporcional al descenso del biomarcador, por primera vez en seis décadas habrá un fármaco aprobado para bajar la Lp(a). Es la próxima revisión editorial obligatoria de esta pieza.
Asociación robusta sin causalidad: HDL-C, homocisteína y testosterona.
Tres casos cerrados en el último decenio, los tres con el mismo patrón: epidemiología positiva, fármacos que mueven el biomarcador sin mover eventos, aleatorización mendeliana que descarta la causalidad simple.
HDL-C: el caso de manual
El «colesterol bueno» asociaba inversamente con eventos, y eso generó tres décadas de fármacos para subirlo. Todos fallaron: la niacina (AIM-HIGH y HPS2-THRIVE detenidos o sin beneficio) y los inhibidores de CETP (torcetrapib aumentó la mortalidad, dalcetrapib falló). La aleatorización mendeliana confirmó que las variantes que suben el HDL-C aisladamente no reducen el infarto. El HDL-C es marcador de salud metabólica general, no protector causal. No se persiguen intervenciones específicas para elevarlo.
Homocisteína: bajar el biomarcador no salva vidas
Seis ensayos aleatorizados grandes (VISP, HOPE-2, NORVIT, WAFACS, VITATOPS, SEARCH), más de 38.000 pacientes, bajaron la homocisteína con ácido fólico y vitaminas B sin reducir eventos. El metaanálisis de aleatorización mendeliana de Clarke (2012) concluyó que la elevación moderada de por vida tiene poco o ningún efecto sobre la cardiopatía coronaria.[12] Caso cerrado, en la pieza sobre homocisteína.
Testosterona y mortalidad: marcador de salud sistémica
La testosterona baja asocia con mortalidad en cohortes, pero la aleatorización mendeliana no respalda la testosterona endógena alta como protectora, y TRAVERSE (2023, n=5.246), el mayor ensayo de terapia sustitutiva, demostró no inferioridad cardiovascular pero no redujo la mortalidad.[13] Es marcador de salud sistémica subyacente, no factor causal aislado, en la pieza sobre testosterona y mortalidad. La lección común a los tres: no todo biomarcador asociado con el riesgo es una diana, y confundirlos genera gasto, prescripción inútil y distracción de lo que sí importa.
SCORE2 y los cálculos a 10 años se quedan cortos; en 2026 las guías de EE.UU. ya lo corrigen.
Los calculadores de riesgo a 10 años fueron útiles para decidir en poblaciones maduras, pero subestiman el riesgo del adulto joven con biomarcadores adversos y omiten las dianas causales. El cambio de 2026 es que las guías han empezado a corregirlo.
SCORE2: qué incluye y qué omite
SCORE2, recomendado por la ESC desde 2021, integra edad, sexo, tabaquismo, presión y colesterol no-HDL, y da un riesgo a 10 años.[11] Lo que no incluye es ApoB, Lp(a), hsCRP, calcio coronario ni marcadores metabólicos. Un varón de 38 años fumador con ApoB de 130 mg/dL puede tener un SCORE2 a 10 años por debajo del 5% y arrastrar tres décadas más de exposición acumulada.
Del retraso a la convergencia 2026
Durante años, EE.UU. fue el rezagado: la guía AHA/ACC de 2018 trataba la Lp(a) y la ApoB como simples «potenciadores de riesgo» opcionales. Eso cambió en marzo de 2026. La guía ACC/AHA/Multisociety de 2026 (Blumenthal, Morris y colaboradores) recomienda por primera vez la medición universal de Lp(a) una vez en la vida con designación Clase I, restaura objetivos de LDL-C y ApoB por categoría de riesgo (ApoB alineada con LDL en <55, <70 y <90 mg/dL), sustituye las ecuaciones Pooled Cohort por las nuevas PREVENT-ASCVD y señala que el cálculo Martin/Hopkins reduce la discordancia frente a Friedewald.[5] EE.UU. y Europa convergen. El consenso ya no está en disputa; lo que falta es la implementación en la consulta.
Lifetime risk: la métrica que cambia decisiones
El riesgo a lo largo de la vida estima la probabilidad de evento antes de los 85-90 años si la exposición actual se mantiene. En adultos jóvenes con biomarcadores adversos puede superar el 50% mientras el riesgo a 10 años queda por debajo del 5%. Esa diferencia es decisiva: el modelo a 10 años recomienda esperar; el modelo de por vida recomienda intervenir pronto. La biología respalda al segundo, porque la variable causal es la exposición acumulada a ApoB, no el riesgo en una ventana arbitraria.
El movimiento Beyond LDL
El cambio lo impulsa una generación de investigadores cardiovasculares: Allan Sniderman (ApoB como diana primaria), Brian Ference (causalidad del LDL), Paul Ridker (la vía inflamatoria, CANTOS), Sam Tsimikas y Benoit Arsenault (Lp(a)). KRECE se posiciona en esta línea: el editorial ancla y los siete satélites son la síntesis en castellano del paradigma Beyond LDL. El estado de adopción por sociedades, tras la convergencia de 2026:
| Sociedad / país | Posición sobre ApoB / Lp(a) | Estado |
|---|---|---|
| EAS / ESC | ApoB preferida en diabetes, obesidad y dislipemia mixta; Lp(a) universal una vez en la vida | Líder |
| ACC / AHA (EE.UU.) | Guía 2026: Lp(a) universal Clase I, objetivos de ApoB, ecuaciones PREVENT | Convergencia 2026 |
| CCS (Canadá) | ApoB superior a LDL-C, uso rutinario recomendado | Adoptado |
| Nórdicas | ApoB en hipercolesterolemia familiar y prevención agresiva | Adoptado |
| NICE (Reino Unido) | Inclusión progresiva basada en UK Biobank | En transición |
| Sociedades españolas | Alineadas con ESC en guías; práctica clínica heterogénea | Transición irregular |
La resistencia a la insulina es el motor que precede a todo.
La conexión entre resistencia a la insulina y enfermedad cardiovascular no es coincidencia epidemiológica: es secuencia mecánica. Es el motor que fabrica las partículas y prepara la pared donde se depositan.
De la VLDL hepática a la ApoB elevada
El hígado con hiperinsulinemia produce más VLDL ricas en triglicéridos, que se convierten en remanentes, IDL y finalmente LDL pequeñas y densas, las más aterogénicas. La ApoB sube porque hay más partículas, aunque el LDL-C siga en rango: es exactamente el patrón de discordancia del 75% en síndrome metabólico. La fisiopatología completa y su precedencia temporal sobre la diabetes están en el cornerstone sobre resistencia a la insulina.
Glicación, disfunción endotelial y placa vulnerable
La hiperglucemia crónica genera productos de glicación avanzada que dañan el glicocálix endotelial y aumentan la permeabilidad arterial, lo que facilita la entrada y la retención de partículas ApoB. El resultado es un paciente que produce más partículas, las hace más dañinas y tiene una arteria más vulnerable a recibirlas. Tratar este eje con fármacos sin corregir la base metabólica es trabajar contra el motor.
La jerarquía KRECE: el motor antes que la farmacología
La jerarquía que KRECE defiende en todo el cluster es explícita: corregir la base metabólica antes de escalar la farmacología. Empezar por las palancas de mayor impacto sistémico (ejercicio de fuerza y resistencia, composición corporal, control de carbohidratos), medir el impacto sobre HOMA-IR, HbA1c, TG/HDL y ApoB a los 3-6 meses, y reservar la intensificación para el riesgo residual real. La estatina, el PCSK9, la natoquinasa y eventualmente el anti-Lp(a) entran cuando están indicados, no antes.
Preguntas frecuentes
¿Qué es la prevención cardiovascular de precisión?
Es un enfoque que sustituye el cálculo de riesgo poblacional a 10 años por tres cosas: estratificación individual con biomarcadores de causalidad genética validada (ApoB, Lp(a)) e imagen subclínica (calcio coronario); intervenciones de mecanismo causal demostrado, es decir, modificar solo lo que ha probado reducir eventos y no solo un biomarcador intermedio; y un horizonte temporal de toda la vida, no de 10 años. El objetivo es tratar antes y mejor al paciente concreto, no al promedio de la población.
¿Qué es la aleatorización mendeliana?
Es un método que usa variantes genéticas, repartidas al azar en la concepción, como un ensayo natural que dura toda la vida. Como esas variantes son independientes del estilo de vida y actúan desde el nacimiento, permiten saber si una exposición (por ejemplo, un LDL alto de por vida) realmente causa la enfermedad, evitando la confusión y la causalidad inversa de los estudios observacionales. Es la herramienta que demostró que el LDL y la Lp(a) son causales, y la que descartó el HDL-C, la homocisteína y la testosterona como dianas modificables.
¿Es mejor medir la ApoB o el colesterol LDL?
La ApoB es mejor predictor: cuenta el número de partículas aterogénicas, mientras que el LDL-C estima la masa de colesterol y puede aparecer normal cuando hay muchas partículas pequeñas y densas (lo habitual en la resistencia a la insulina). La ApoB está estandarizada y no depende del ayuno. Donde se pueda medir, es la métrica de elección para decidir y seguir el tratamiento.
¿Qué es el riesgo residual cardiovascular?
Es la probabilidad de evento que persiste pese a tener el LDL-C en rango con estatinas. Como la reducción total que se logra bajando el LDL rara vez supera el 30%, queda un riesgo grande que se reparte en cuatro componentes: aterogénico (partículas que el LDL-C no cuenta), inflamatorio (hsCRP), trombótico (fibrinólisis) y metabólico (resistencia a la insulina). Cada uno se mide y se trata de forma distinta.
¿Es fiable calcular el riesgo cardiovascular a 10 años con SCORE2?
Es útil para decidir en personas maduras, pero se queda corto en adultos jóvenes con biomarcadores adversos y omite la ApoB, la Lp(a), la hsCRP y el calcio coronario. La métrica que importa en longevidad es el riesgo a lo largo de la vida: alguien de 40 años con ApoB alta puede tener un riesgo a 10 años bajo y un riesgo de por vida superior al 50%. Por eso conviene complementarlo con biomarcadores causales y, si procede, imagen subclínica.
¿El HDL alto protege el corazón?
No de forma causal. El HDL-C alto asocia con menos eventos, pero los fármacos que lo suben (niacina, inhibidores de CETP) no han reducido el riesgo, y la aleatorización mendeliana confirma que subirlo de forma aislada no protege. El HDL-C es un marcador de salud metabólica general, no una diana que tratar. Perseguir un HDL alto con suplementos o fármacos no está respaldado por la evidencia.
- Sniderman AD, Thanassoulis G, Glavinovic T, Navar AM, Pencina M, Catapano A, Ference BA. Apolipoprotein B Particles and Cardiovascular Disease: A Narrative Review. JAMA Cardiol. 2019;4(12):1287-1295. PubMed. Superioridad de la ApoB sobre LDL-C, no-HDL-C y LDL-P.
- Hill AB. The Environment and Disease: Association or Causation? Proc R Soc Med. 1965;58(5):295-300. PubMed. Los nueve criterios para juzgar causalidad a partir de una asociación.
- Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38(32):2459-2472. PubMed. La sentencia causal sobre el LDL.
- Kronenberg F, Mora S, Stroes ESG, et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: a European Atherosclerosis Society consensus statement. Eur Heart J. 2022;43(39):3925-3946. PubMed. Consenso EAS/ESC: medir Lp(a) una vez en la vida; umbrales.
- Blumenthal RS, Morris PB, Gaudino M, et al. 2026 ACC/AHA/Multisociety Guideline on the Management of Dyslipidemia. J Am Coll Cardiol. Publicado online 13 marzo 2026. doi:10.1016/j.jacc.2025.11.016. JACC. Medición universal de Lp(a) (Clase I), objetivos de ApoB, ecuaciones PREVENT-ASCVD, Martin/Hopkins.
- Nordestgaard BG. Triglyceride-Rich Lipoproteins and Atherosclerotic Cardiovascular Disease: New Insights From Epidemiology, Genetics, and Biology. Circ Res. 2016;118(4):547-563. PubMed. Colesterol remanente y partículas ricas en triglicéridos como riesgo residual.
- Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease (CANTOS). N Engl J Med. 2017;377(12):1119-1131. PubMed. Confirmación causal de la vía inflamatoria sin modificar LDL-C.
- Sabatine MS, Giugliano RP, Keech AC, et al; FOURIER Steering Committee and Investigators. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med. 2017;376(18):1713-1722. PubMed. PCSK9: reducción de eventos con outcomes duros.
- Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence Variations in PCSK9, Low LDL, and Protection against Coronary Heart Disease. N Engl J Med. 2006;354(12):1264-1272. PubMed. Validación genética de PCSK9 como diana.
- Nicholls SJ, Nissen SE, Fleming C, et al. Muvalaplin, an Oral Small Molecule Inhibitor of Lipoprotein(a) Formation: A Randomized Clinical Trial. JAMA. 2023;330(11):1042-1053. PubMed. Inhibidor oral del ensamblaje de Lp(a); contexto del pipeline.
- Visseren FLJ, Mach F, Smulders YM, et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur Heart J. 2021;42(34):3227-3337. PubMed. SCORE2 y el marco europeo de prevención.
- Clarke R, Bennett DA, Parish S, et al. Homocysteine and coronary heart disease: meta-analysis of MTHFR case-control studies, avoiding publication bias. PLoS Med. 2012;9(2):e1001177. PubMed. La aleatorización mendeliana descarta la homocisteína como causa.
- Lincoff AM, Bhasin S, Flevaris P, et al; TRAVERSE Study Investigators. Cardiovascular Safety of Testosterone-Replacement Therapy. N Engl J Med. 2023;389(2):107-117. PubMed. No inferioridad cardiovascular, sin reducción de mortalidad.