La mitofagia cerebral no es solo eliminar las mitocondrias dañadas: es fabricar las nuevas al mismo tiempo. La rapamicina hace las dos cosas a la vez, y de ahí nace la hipótesis de que podría frenar el Alzheimer.
La narrativa preclínica es real y está bien construida. Pero la traducción clínica en humanos acaba de chocar con su primer dato real, y obliga a recalibrar el discurso.
El primer ensayo Fase 1 en humanos con deterioro cognitivo encontró que, a dosis tolerable, la rapamicina oral no llega medible al cerebro. Este artículo ordena la mitad de la evidencia preclínica del campo y la totalidad de los datos humanos verificados, sin endulzar.
Esta es la pieza del lado neuronal del control de calidad mitocondrial. El concepto general de fabricar mitocondrias nuevas lo tratamos en el pilar de biogénesis mitocondrial, y el lado muscular y nutricional, en la pieza sobre mitofagia, ejercicio y urolitina A. Aquí cubrimos el cerebro, donde la regla cambia: la neurona no se divide, no puede diluir el daño, y depende críticamente de reciclar sus orgánulos defectuosos.
Mitofagia neuronal: el control de calidad que el cerebro no puede delegar
La mitofagia es un proceso de macroautofagia selectiva: la célula identifica, marca y degrada mitocondrias individuales que han perdido potencial de membrana, sobreproducen ROS o tienen defectos en la cadena respiratoria. No es autofagia general, no se engullen porciones aleatorias del citoplasma. Se elige específicamente la mitocondria dañada para sacrificarla.
Mitofagia frente a autofagia: la diferencia que casi todos confunden
La autofagia general responde a demanda: la célula tiene hambre y recicla lo que pilla. La mitofagia responde a oferta: una mitocondria concreta se ha dañado y se ofrece para degradación. Ambas comparten la maquinaria final (autofagosoma y lisosoma), pero las señales de inicio son distintas: privación de nutrientes frente a despolarización mitocondrial [1].
Mitofagia neuronal frente a muscular: por qué no son intercambiables
Esta distinción es la que más se pasa por alto. El músculo activa mitofagia sobre todo vía AMPK→ULK1 ante estrés energético agudo: la contracción consume ATP, sube el ratio AMP/ATP, AMPK fosforila ULK1 y se dispara el autofagosoma sobre la mitocondria dañada [2]. En la neurona post-mitótica, en cambio, la vía dominante es PINK1-Parkin: ubiquitinación directa de la membrana mitocondrial externa, reconocimiento por adaptadores como P62 y NDP52, y degradación lisosomal. La mutación de PINK1 o Parkin causa Parkinson de inicio temprano en humanos (evidencia causal de que la mitofagia neuronal no es opcional), mientras que esas mismas mutaciones en músculo apenas dan fenotipo [3].
Por qué el cerebro depende de la mitofagia más que ningún órgano
Cuatro razones objetivas. Una, el cerebro consume el 20-25 % del oxígeno corporal con solo el 2 % de la masa: cualquier ineficiencia mitocondrial se nota antes ahí. Dos, las neuronas no se dividen, no pueden diluir mitocondrias dañadas repartiéndolas entre células hijas como hace un hepatocito. Tres, la conexión sináptica depende de un suministro local de ATP en terminales axonales que pueden estar muy lejos del cuerpo celular, lo que hace crítica la calidad de cada mitocondria local; el deterioro de la mielina con la edad agrava esa vulnerabilidad axonal. Cuatro, la microglía vecina no tolera mitocondrias neuronales fugando contenido oxidado: las interpreta como señal de daño y activa una respuesta inflamatoria que daña a las neuronas sanas adyacentes.
Qué pasa cuando la mitofagia falla en el cerebro
Cuando el flujo mitofágico se detiene, ocurren cinco cosas en cascada: se acumulan mitocondrias defectuosas en el soma y las terminales, cae la producción local de ATP, sube el estrés oxidativo crónico, se activa la vía apoptótica mitocondrial por liberación de citocromo c, y la microglía adopta un fenotipo proinflamatorio sostenido. Este es el bucle que conecta disfunción mitocondrial con Alzheimer, Parkinson y demencia [4]: no son enfermedades de la mitocondria, son enfermedades de un sistema de control de calidad que ha fallado. Las cuatro vías principales de mitofagia neuronal (PINK1-Parkin, dependiente de ubiquitina y dominante en neurona; BNIP3 y NIX; FUNDC1; y FKBP8) convergen en el mismo final. La rapamicina no actúa sobre ellas directamente: las habilita aguas abajo al inhibir mTORC1, que en abundancia frena ULK1 y bloquea el inicio del autofagosoma.
El control de calidad mitocondrial tiene dos caras, no una
El error típico al hablar de mitofagia es presentarla como el proceso completo. No lo es. La mitofagia es la mitad, la destructiva, de un sistema con dos caras inseparables. Sin la otra mitad, la biogénesis, eliminar mitocondrias defectuosas sin fabricar nuevas dejaría a la neurona desabastecida.
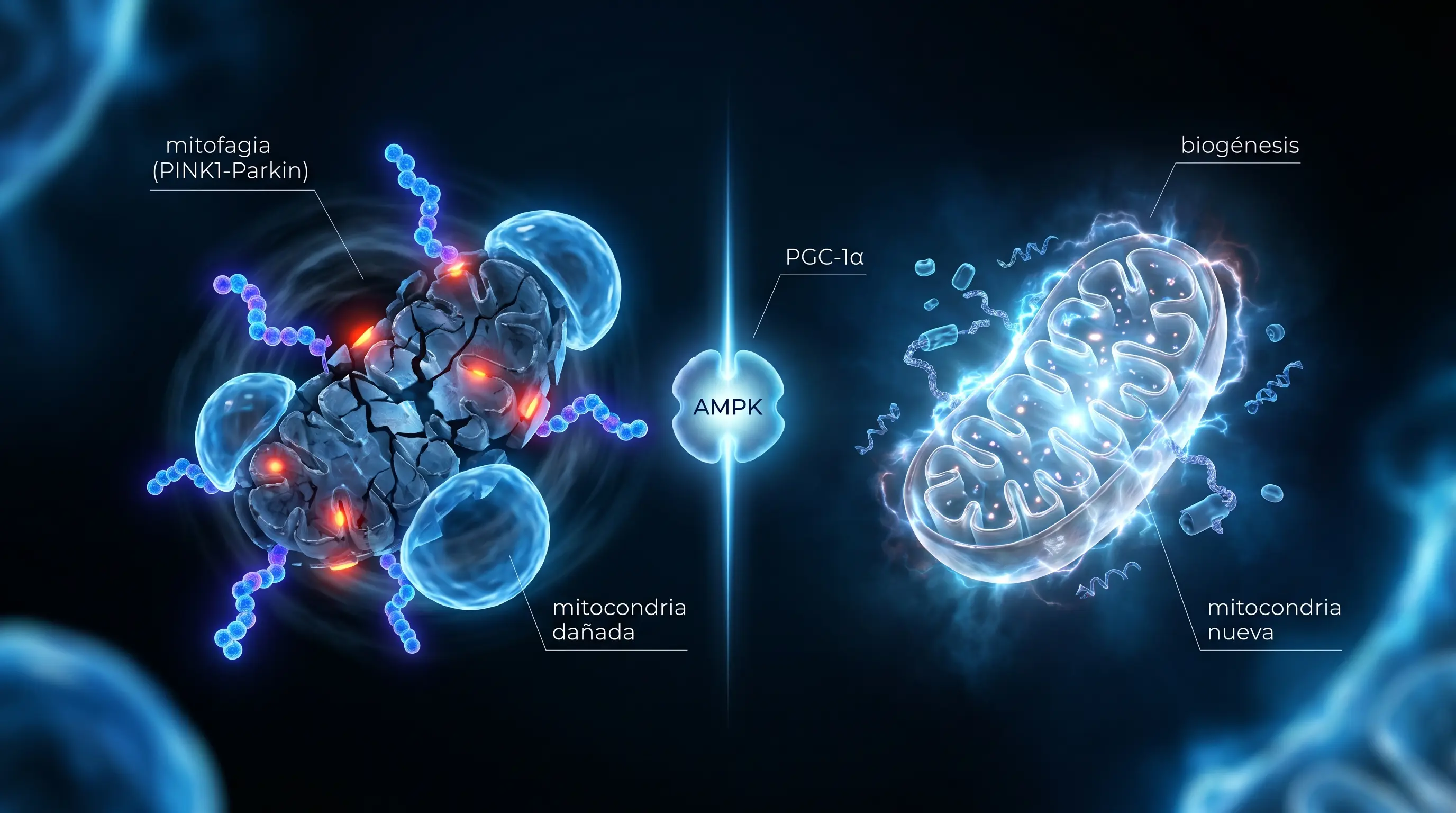
La cara que elimina
La mitocondria que pierde potencial de membrana estabiliza PINK1 en su superficie. PINK1 fosforila ubiquitina y recluta a Parkin, una E3 ubiquitin-ligasa que decora la mitocondria con cadenas de poliubiquitina. Los adaptadores P62, OPTN y NDP52 reconocen esas marcas y reclutan LC3B, la proteína que define la membrana del autofagosoma, envolviendo la mitocondria. El autofagosoma se fusiona con el lisosoma, el contenido se degrada y los aminoácidos y nucleótidos reciclados vuelven al pool celular [1].
La cara que fabrica, en el cerebro
La biogénesis mitocondrial es la fabricación de mitocondrias nuevas, y su regulador maestro es PGC-1α; el mecanismo completo (NRF1, NRF2, TFAM y la coordinación de los dos genomas) lo desarrollamos en el pilar de biogénesis. Lo específico del cerebro es que PGC-1α declina con la edad en el tejido nervioso y está disminuido en el hipocampo y la corteza de pacientes con Alzheimer. Y aquí la biogénesis no es solo reemplazo cuantitativo, es cualitativo: las mitocondrias nuevas tienen ADN mitocondrial sin mutaciones acumuladas, cadenas respiratorias funcionales y transporte axonal preservado. En el músculo, el inductor fisiológico más potente es el ejercicio aeróbico; en el cerebro añade la señal del lactato circulante.
Por qué solo limpiar (o solo fabricar) no funciona
Una neurona que solo elimina mitocondrias acabaría sin reserva energética. Una que solo fabrica acabaría llena de mitocondrias defectuosas. El balance se llama turnover mitocondrial, y es justo lo que el cerebro envejecido pierde: ralentiza la mitofagia, ralentiza la biogénesis y mantiene mitocondrias viejas funcionando peor.
El nodo que coordina las dos caras: AMPK
La elegancia bioquímica está en que el mismo sensor, AMPK, activa los dos lados a la vez. AMPK fosforila ULK1 para iniciar la mitofagia y, en paralelo, activa SIRT1 (subiendo NAD+), que deacetila a PGC-1α y dispara el programa de biogénesis [5]. Una señal, dos procesos coordinados. Por eso el ejercicio funciona: AMPK sube y ambos lados se mueven juntos. A esto se suman la fisión (DRP1, FIS1), que fragmenta lo dañado para que el autofagosoma pueda engullirlo, y la fusión (OPA1, MFN1/2); sin fisión previa no hay mitofagia eficiente, y en el cerebro envejecido también se ralentiza.
Rapamicina y mTOR: qué es, cómo actúa, qué nombres lleva
Rapamicina, sirolimus y rapálogos: el mismo fármaco con varios nombres
La rapamicina es un macrólido aislado en 1972 de una bacteria del suelo (Streptomyces hygroscopicus) en la Isla de Pascua, Rapa Nui, de ahí el nombre. Su nombre como medicamento es sirolimus (Rapamune). Modificada químicamente da la familia de los rapálogos: everolimus (RAD001), temsirolimus, ridaforolimus. En la literatura geroprotectora aparecen indistintamente; los nombres de prescripción en EE. UU. y Europa son sirolimus y everolimus. Más contexto en el Bio de rapamicina de KRECE.
Cómo inhibe mTORC1 y por qué eso activa la mitofagia
La rapamicina se une a una proteína intracelular, FKBP12, formando un complejo que inhibe alostéricamente a mTORC1. En estado basal, mTORC1 fosforila ULK1 en sitios inhibitorios y bloquea el inicio de la autofagia, porque la célula tiene nutrientes y no necesita reciclar. Cuando la rapamicina apaga mTORC1, ULK1 queda libre para que AMPK la active y la maquinaria del autofagosoma arranca [3]. A esto se suma que inhibir mTORC1 reorganiza la máquina traduccional hacia el programa de proteostasis.
mTORC1 frente a mTORC2: qué se inhibe y qué se preserva
mTOR existe en dos complejos. mTORC1 regula crecimiento y anabolismo y bloquea la autofagia: es el target de la rapamicina y el que se quiere inhibir. mTORC2 regula supervivencia, citoesqueleto y metabolismo de la insulina: su inhibición prolongada produce los efectos metabólicos clásicos (hiperglucemia, dislipidemia, resistencia a la insulina). La rapamicina no se une directamente a mTORC2, pero la administración crónica diaria desestabiliza progresivamente sus complejos [6]. De ahí la estrategia geroprotectora moderna de dosis intermitente semanal: pulso suficiente para inhibir mTORC1, valle suficiente para preservar mTORC2.
El mTOR de la neurona no es el del músculo
En el músculo, la activación pulsátil de mTORC1 es deseable: dispara síntesis proteica y crecimiento, y por eso la rapamicina crónica preocupa a quien busca hipertrofia. En la neurona post-mitótica el cálculo es distinto: no hay hipertrofia que perseguir y sí hay agregados proteicos que limpiar. Inhibir parcialmente mTORC1 favorece la proteostasis (el reciclaje de proteínas mal plegadas como amiloide-β y tau hiperfosforilada) sin un coste anabólico equivalente. Eso justifica por qué el cerebro es un órgano diana para rapamicina aunque el músculo no lo sea igual.
¿La rapamicina cruza la barrera hematoencefálica?
Aquí la narrativa preclínica colisiona con la farmacocinética humana. En modelos animales con dosis altas y crónicas, la rapamicina alcanza concentraciones medibles en parénquima cerebral e inhibe mTORC1 en hipocampo y corteza [7]. En humanos a dosis geroprotectoras el escenario es muy distinto: el Fase 1 piloto Gonzales 2025, en Communications Medicine, midió rapamicina por espectrometría de masas en líquido cefalorraquídeo antes y después de 8 semanas a 1 mg/día en 10 pacientes con deterioro cognitivo leve o Alzheimer. La rapamicina no fue detectable en el LCR en ningún momento [8]. El dato obliga a separar dos hipótesis que se trataban como una: efecto cerebral directo por inhibición de mTORC1 neuronal, o efecto indirecto por modulación periférica de la inflamación y la inmunidad.
Lo que la nueva investigación revela sobre rapamicina y mitocondria cerebral
La hipótesis de que la rapamicina coordina ambos lados del control de calidad en cerebro vulnerable descansa en tres papers preclínicos. Vamos uno a uno, con la fuente primaria delante.
Mitofagia: Wang et al. 2021 en modelo de Alzheimer
Wang y colaboradores publicaron en The Journals of Gerontology: Series A (2021) un estudio en ratones APP/PS1, el modelo transgénico clásico de Alzheimer, tratados con rapamicina 8 semanas [9]. Los hallazgos verificados: aumento de mitofagia Parkin-mediada en el hipocampo (co-localización de TOM20 con LC3B), fusión completa mitofagosoma-lisosoma, mejora de aprendizaje, memoria y plasticidad sináptica, más proteínas sinápticas, menos apoptosis por citocromo c y recuperación parcial de la función mitocondrial. La cifra de «+400 %» que circula en divulgación es una interpretación de las figuras, no una magnitud del abstract; lo que queda en firme es la dirección del efecto y su asociación con mejora cognitiva.
Biogénesis: Reid et al. 2020 y la trampa de generalizar
El grupo de Benjamin Miller publicó en 2020 el primer estudio que mide directamente la síntesis de proteínas mitocondriales cerebrales con rapamicina, usando agua deuterada como trazador en ratones UM-HET3 (el del ITP del NIA), jóvenes y viejos, 8 semanas [10]. Los hallazgos son más matizados que la versión popularizada: en machos jóvenes, sin efecto; en hembras jóvenes, la combinación con metformina disminuyó la síntesis; y solo en la cohorte vieja (que en el diseño final solo incluyó hembras) la rapamicina aumentó significativamente la síntesis mitocondrial cerebral.
Cuidado con generalizar Reid 2020. El hallazgo correcto, en palabras de los autores, es que «las respuestas a tratamientos de envejecimiento lento varían con sexo y edad». La rapamicina sube la biogénesis mitocondrial cerebral solo en hembras viejas, no en machos viejos (no se pudieron medir), no en machos jóvenes y no en hembras jóvenes. Saltar a «la rapamicina aumenta la biogénesis cerebral en el envejecimiento» es ir más allá de lo que los datos sostienen.
El mecanismo dual: limpiar y fabricar a la vez
Tomados juntos, Wang 2021 y Reid 2020 son la base mecánica de la afirmación: la rapamicina activa mitofagia y biogénesis en cerebro, en paralelo. Es coherente, al inhibir mTORC1 se libera ULK1 para la mitofagia y se reduce a la vez el freno sobre PGC-1α, permitiendo reponer lo que se degrada. La narrativa preclínica está bien construida.
Por qué solo funciona en cerebro envejecido
El dato más contraintuitivo y el más relevante: el efecto sobre biogénesis aparece solo en la cohorte vieja. La interpretación más plausible es que en cerebro joven la mitofagia y la biogénesis funcionan bien y mTOR no es el cuello de botella; en cerebro envejecido, donde ambos procesos están ralentizados, inhibir mTORC1 libera los dos detenidos. Es un mecanismo de rescate de función perdida, no de optimización de función preservada. Implica que usar rapamicina «preventiva» en un cerebro de 35 años carece de soporte mecánico en estos datos.
Drake 2021 y Mito 2022: dos avisos sobre lo muscular y lo cerebral
Drake y colaboradores (PNAS, 2021) mostraron que AMPK no solo actúa en el citosol: se transloca físicamente a la membrana mitocondrial tras estrés energético y acelera la mitofagia local, y que la metformina induce el mismo mecanismo [11]. El hallazgo es muscular, no cerebral, y abre la pregunta de si opera igual en neurona, todavía sin replicar. El estudio de Mito y colaboradores (Cell Metabolism, 2022) en ratones Deletor mostró que la mitofagia muscular es heterogénea (en mosaico) y que la rapamicina restaura el flujo en las fibras detenidas, pero el modelo es de enfermedad mitocondrial muscular, no de envejecimiento cerebral, y los autores no extrapolan al cerebro [12]. Trasladar Mito 2022 al contexto cerebral, común en divulgación, es un salto que los datos no sostienen.
Rapamicina y Alzheimer: dónde está la evidencia humana real
Hasta aquí la narrativa preclínica es sólida. Lo que sigue es donde ese discurso colisiona con la traducción clínica, y donde un buen lector debería recalibrar expectativas.
La hipótesis Kaeberlein-Galvan 2019: por qué pidieron un ensayo
En enero de 2019, Matt Kaeberlein y Veronica Galvan publicaron en Science Translational Medicine un Perspective titulado «Rapamycin and Alzheimer’s disease: Time for a clinical trial?» [13]. El argumento: la rapamicina tiene un expediente preclínico extraordinario en modelos de Alzheimer (reduce amiloide-β y tau, restaura el flujo cerebral, preserva la barrera hematoencefálica, mejora cognición), está aprobada por la FDA desde 1999 y su seguridad está caracterizada tras décadas de uso en trasplante; y sin embargo ningún ensayo la había probado en Alzheimer. El obstáculo que señalan no es científico sino económico: la rapamicina es genérica, no patentable, y nadie financia un Fase 3 de decenas de millones para un fármaco que no podrá comercializar con margen.
La evidencia preclínica y la espada de doble filo
Lo que apilaron como justificación incluye múltiples modelos murinos (3xTg-AD, hAPP/J20, APP/PS1, PDAPP) con reducción de amiloide soluble e insoluble, menos tau hiperfosforilada y ovillos, restauración del flujo cerebral, preservación de la barrera y mejoras cognitivas funcionales. La respuesta académica llegó rápido: Carosi y Sargeant publicaron en Autophagy ese mismo año «Rapamycin and Alzheimer disease: a double-edged sword?» [14], matizando que inducir autofagia solo beneficia si el sistema lisosomal aguas abajo funciona; en Alzheimer avanzado, con lisosomas sobrecargados, hiperactivar la autofagia sin descarga podría empeorar la patología. La ventana terapéutica, sugieren, estaría en fases tempranas (deterioro cognitivo leve), no en Alzheimer establecido.
El dato que cambia el discurso: el Fase 1 piloto Gonzales 2025
En mayo de 2025, Mitzi Gonzales y colaboradores (Glenn Biggs Institute, UT Health San Antonio, con Galvan como coautora) publicaron en Communications Medicine el primer Fase 1 humano diseñado para responder a esa pregunta [8].
| Parámetro | Valor |
|---|---|
| Tipo de estudio | Abierto, un solo centro, Fase 1 piloto |
| Población | 10 participantes, edad media 74 ± 4 años, 60 % mujeres |
| Diagnóstico | Deterioro cognitivo leve o demencia leve por Alzheimer |
| Intervención | Rapamicina (sirolimus) 1 mg/día oral, 8 semanas |
| Endpoint primario | Penetración en el SNC por espectrometría de masas en LCR, antes y después |
| Endpoints secundarios | Seguridad, cognición, biomarcadores de Alzheimer e inflamatorios en LCR y plasma |
| Endpoint | Resultado | Dirección |
|---|---|---|
| Rapamicina en LCR | No detectable antes ni después | No penetra |
| Cambios cognitivos | Ninguno significativo | Nulo |
| HbA1c | Aumentó | Empeoró |
| Presión sistólica | Aumentó | Empeoró |
| p-tau-181 en LCR | +2,64 pg/ml (IC 95 % 0,70-4,59) | Aumentó |
| GFAP en LCR | +6262 pg/ml (IC 95 % 3787-9374) | Aumentó |
| NfL en LCR | +367 pg/ml (IC 95 % 204-562) | Aumentó |
| Eventos adversos | 20 totales, mayoría leves | Tolerable |
Los autores son los primeros en reconocer la ambigüedad: el aumento de p-tau-181, GFAP y NfL en LCR podría reflejar empeoramiento de la enfermedad o mayor depuración de proteínas al activarse parcialmente la autofagia, y con 8 semanas y 10 pacientes es imposible discriminar. Lo que queda en firme: con 1 mg/día durante 8 semanas, la rapamicina no llega medible al LCR, no produce cambios cognitivos detectables y añade algo de coste metabólico.
Qué obliga a recalibrar
Gonzales 2025 recalibra el discurso en tres direcciones. Una, si el efecto cerebral existe en humanos, no parece pasar por concentraciones parenquimatosas medibles a esta dosis: o el mecanismo es periférico, o requiere dosis que la seguridad no permite. Dos, 8 semanas pueden ser insuficientes frente a tratamientos de meses en modelos murinos; el nuevo ensayo lanzado en marzo de 2026 por la UT Health San Antonio (n=84, financiado por el NIA, foco en dosis y seguridad en mayores sanos) dará pistas [15]. Tres, la posición responsable hoy es que la rapamicina para prevenir o tratar Alzheimer es una hipótesis con respaldo preclínico sólido y un primer dato humano sobrio, no una intervención validada. Más contexto en el análisis de rapamicina, ITP y PEARL.
Mannick 2014: la prueba humana más sólida sobre mTOR e inmunidad
Para no cerrar con un solo dato negativo, conviene completar la foto. El ensayo de Joan Mannick (Novartis, 2014) en Science Translational Medicine es la mejor evidencia humana de que inhibir parcialmente mTORC1 es seguro y eficaz para algo medible: 218 mayores de 65 años, everolimus a dosis bajas durante 6 semanas, seguido de vacuna antigripal [16]. Resultados: respuesta a la vacuna mejorada ~20 % con las dosis bajas, reducción de PD-1 en linfocitos T (marcador de inmunosenescencia), buena seguridad y sin alteración metabólica relevante (la pauta intermitente preserva mTORC2). La continuación de 2018 confirmó menos infecciones respiratorias en 264 mayores [17]. La evidencia humana sólida sobre mTOR en envejecimiento existe, pero es para inmunidad, no para cognición. Ese matiz importa.
Cómo se mide la mitofagia cerebral (y por qué casi no se mide en humanos)
La mitofagia es un proceso dinámico. La fotografía estática de una biopsia o una muestra de LCR captura, en el mejor de los casos, marcadores indirectos. El proceso real solo se ve con tejido vivo o reportero fluorescente: modelos animales sí, humanos no.
Modelos animales: MitoQC, MitoKeima y MitoTimer
| Sistema | Mecanismo | Ventaja | Limitación |
|---|---|---|---|
| MitoQC | mCherry-GFP en membrana externa; neutro amarillo, lisosoma ácido solo rojo | Cuantifica el evento de degradación | Solo animales transgénicos |
| MitoKeima | Proteína resistente a proteasas con excitación pH-dependiente | Bimodal muy sensible | Se destruye con fijación; solo tejido fresco |
| MitoTimer | Verde (sintetizada) a rojo (oxidada/madura); mide edad mitocondrial | Sensible a estrés oxidativo | Mide turnover global, no eventos de mitofagia |
Biomarcadores indirectos en humanos
En humanos se mide expresión y abundancia de proteínas implicadas (PINK1, Parkin, P62/SQSTM1, LC3B-II, ULK1, FUNDC1) por western blot o PCR en biopsias musculares o muestras post-mórtem. La limitación crítica es que son snapshots: decir «aumentó la mitofagia» porque sube LC3B-II en biopsia es decir «la maquinaria está más presente», no «el proceso funciona más». Son cosas distintas.
Por qué medirla en una persona viva sigue siendo imposible
El cerebro humano vivo no se biopsia. La única forma de medir mitofagia cerebral en humanos hoy es post-mórtem, lo que da cerebros enfermos al final de la enfermedad, el peor punto del proceso. Las técnicas de imagen disponibles (PET de glucosa, amiloide o tau, DTI de materia blanca) no miden mitofagia, miden consecuencias aguas abajo. El campo necesita un trazador PET específico de flujo mitofágico y nadie lo tiene. Por eso el endpoint primario de Gonzales 2025 fue la penetración del fármaco en LCR, no la mitofagia: nadie sabe medirla en humanos vivos.
Lo que se infiere desde biomarcadores periféricos
Algunos estudios miden proxies periféricos: p-tau-181 (tau patológica), GFAP (gliosis), NfL (daño axonal), Aβ42/40 (carga amiloide). El supuesto es que si la mitofagia limpia agregados, esos marcadores deberían bajar con tiempo. Gonzales 2025 los midió en LCR y los vio subir a 8 semanas, lo que admite dos lecturas contrarias: empeoramiento de la enfermedad, o aumento transitorio de turnover proteico que volvería a basal. Sin brazo placebo y con n=10, la inferencia se queda en hipótesis. Es la clase de pregunta que solo el ensayo de 84 personas puede empezar a responder.
Otros inductores de mitofagia cerebral más allá de la rapamicina
La rapamicina no es la única palanca. Y en humanos, hoy, no es ni la más respaldada por datos cognitivos. Aquí van las cinco con base mecánica relevante para cerebro, en orden de evidencia humana.
Ejercicio aeróbico e HIIT: el más potente y mejor documentado
El ejercicio activa AMPK, que fosforila ULK1 y dispara mitofagia, y a la vez activa PGC-1α vía SIRT1 disparando biogénesis. Además genera lactato circulante, que cruza la barrera y actúa como señal de neuroplasticidad (sube BDNF, induce angiogénesis cerebral, mejora flujo). En mayores, 12 meses de entrenamiento aeróbico moderado-vigoroso aumentan el volumen hipocampal en torno a un 2 %, revirtiendo la atrofia anual fisiológica. No hay un solo fármaco con esa evidencia en cerebro humano. Cómo estructurarlo, en el satélite de biogénesis y ejercicio.
Urolitina A: datos musculares sólidos, cerebrales pendientes
La urolitina A tiene los mejores ensayos humanos para mitofagia muscular: tres RCTs en revistas de primer nivel con 500-1000 mg/día durante 4 meses, con mejoras de fuerza y resistencia [18]. La aplicación cerebral está mucho menos desarrollada: hay datos preclínicos prometedores, pero ningún RCT con endpoint cognitivo primario en humanos. El lado muscular y nutricional, en la pieza sobre mitofagia, ejercicio y urolitina A.
Ayuno prolongado: el dato de Mattson y Longo
El ayuno activa AMPK por caída de glucosa e insulina y eleva cuerpos cetónicos (sobre todo β-hidroxibutirato), que el cerebro usa como combustible y que actúan como señal epigenética subiendo BDNF. En modelos animales (Mattson, NIH; Longo, USC), ayunos de días alternos o protocolos FMD activan mitofagia neuronal y mejoran memoria. En humanos la evidencia es indirecta (marcadores de autofagia en leucocitos tras ayuno, mejoras cognitivas tras FMD en deterioro leve), y la traslación exacta del tiempo de ayuno entre ratón y humano sigue discutida.
Espermidina: el ensayo SmartAge
La espermidina es una poliamina endógena que induce autofagia y mitofagia por inhibición de la acetiltransferasa EP300 [19]. En mayores con quejas subjetivas de memoria, un ensayo piloto de Wirth y colaboradores (3 meses, n=30, dentro del programa SmartAge) administró una dosis baja de espermidina de germen de trigo y documentó mejoras en memoria frente a placebo [20]. Es la mejor señal humana cognitiva de un compuesto autofágico oral hoy, pero procede de un piloto pequeño, con magnitud modesta y pendiente de replicación sólida.
Restricción calórica
La restricción calórica crónica activa AMPK, suprime mTOR e induce un patrón similar al ayuno sostenido. En primates hay dos estudios pivotantes con resultados divergentes (Wisconsin positivo, NIA neutro), lo que indica dependencia de la calidad de la dieta basal y el fondo genético. En humanos sanos, el ensayo CALERIE-2 (restricción del 25 % durante 2 años) documentó mejoras en relojes epigenéticos sin datos cognitivos primarios. La aplicación cerebral sigue siendo inferencial.
Jerarquía de palancas: qué empezar primero
| Intervención | Mecanismo cerebral | Evidencia humana cognitiva | Posición KRECE |
|---|---|---|---|
| Ejercicio aeróbico / HIIT | AMPK→ULK1 + PGC-1α; lactato→BDNF | Alta (RCTs hipocampo) | Base no negociable |
| Espermidina | Inhibición EP300; autofagia general | Moderada (SmartAge) | Suplementación creíble |
| Ayuno / FMD | AMPK alto; BHB señal epigenética | Indirecta | Práctica periódica |
| Urolitina A | Mitofagia selectiva sin inhibir mTOR | Sólida muscular, cerebral pendiente | Esperar datos cognitivos |
| Restricción calórica | AMPK alto; mTOR bajo crónico | Marcadores epigenéticos | No universal |
| Rapamicina low-dose | Inhibición mTORC1; mitofagia + biogénesis | Preclínica fuerte; Fase 1 sobrio | Experimental supervisado |
Preguntas frecuentes
¿Qué es la mitofagia en una frase?
La mitofagia es la eliminación selectiva de mitocondrias dañadas dentro de una célula mediante autofagia, un proceso que mantiene la calidad mitocondrial y evita la acumulación de orgánulos defectuosos.
¿Qué diferencia hay entre mitofagia y autofagia?
La autofagia es el proceso general por el que la célula recicla componentes citoplasmáticos en respuesta a privación de nutrientes. La mitofagia es una forma selectiva de autofagia que elige específicamente mitocondrias dañadas para degradación. Comparten la maquinaria final (autofagosoma y lisosoma), pero las señales de inicio son distintas.
¿Qué es la mitofagia cerebral o neuronal?
Es la mitofagia que ocurre en las neuronas del sistema nervioso central. Es especialmente crítica porque las neuronas son células post-mitóticas (no se dividen) y no pueden diluir mitocondrias dañadas mediante división celular. Su fallo se relaciona con Alzheimer, Parkinson y demencia.
¿Cómo activar la mitofagia cerebral de forma natural?
Las palancas con base mecánica más sólida son el ejercicio aeróbico e HIIT (subir AMPK), el ayuno prolongado (activar AMPK y subir β-hidroxibutirato), la espermidina dietética, la restricción calórica cíclica y el sueño de calidad (limpieza glinfática nocturna).
¿Cómo se mide la mitofagia en humanos?
En la práctica directa, no se puede. Los biomarcadores indirectos disponibles son la expresión de PINK1, Parkin, P62, LC3B y ULK1 en biopsia muscular o post-mórtem. Los modelos de reportero fluorescente (MitoQC, MitoKeima, MitoTimer) son exclusivos de animales transgénicos. Medir mitofagia cerebral en una persona viva sigue siendo imposible.
¿Qué es la rapamicina y para qué sirve?
La rapamicina (sirolimus, Rapamune) es un macrólido producido por Streptomyces hygroscopicus, aprobado por la FDA desde 1999 como inmunosupresor para prevenir el rechazo de trasplantes. Su mecanismo es la inhibición alostérica de mTORC1, lo que activa la autofagia y reduce la síntesis proteica anabólica. Es el geroprotector con el expediente preclínico más sólido del campo.
¿Qué diferencia hay entre rapamicina, sirolimus y everolimus?
Rapamicina y sirolimus son el mismo compuesto; sirolimus es el nombre como medicamento (Rapamune). Everolimus (RAD001) es un rapálogo, una rapamicina modificada químicamente con perfil farmacocinético mejorado, usado en indicaciones oncológicas. Comparten mecanismo y en investigación geroprotectora se usan indistintamente.
¿La rapamicina cruza la barrera hematoencefálica en humanos?
A dosis de 1 mg/día durante 8 semanas, el Fase 1 piloto de Gonzales 2025 demostró que la rapamicina no es detectable en líquido cefalorraquídeo en pacientes con MCI/Alzheimer. En modelos animales sí alcanza concentraciones cerebrales con dosis más altas. La hipótesis actual es que el efecto cerebral en humanos podría ser indirecto (modulación periférica de inflamación e inmunidad), no por inhibición directa de mTORC1 neuronal.
¿Sirve la rapamicina para prevenir el Alzheimer en personas sanas?
A día de hoy, ningún ensayo clínico humano ha demostrado prevención de Alzheimer con rapamicina. La evidencia preclínica es robusta; la traslación humana está en fase muy temprana. El primer Fase 1 piloto en MCI/Alzheimer (Gonzales 2025) no mostró mejora cognitiva a 8 semanas. La respuesta responsable es que no hay evidencia humana para indicar rapamicina como prevención de Alzheimer.
¿La rapamicina debilita el sistema inmune?
Depende de la dosis y la pauta. En dosis altas continuas (trasplante, 2-5 mg/día sostenido) sí: es inmunosupresora. En dosis bajas intermitentes (everolimus 5 mg/semana en Mannick 2014) hace lo contrario: mejoró en torno a un 20 % la respuesta a la vacuna antigripal en mayores y redujo PD-1 en linfocitos T, es decir, mejoró la función inmune envejecida.
¿Qué dosis de rapamicina se usa para longevidad?
No hay una dosis validada por un ensayo clínico humano de longevidad terminado. Las pautas usadas off-label oscilan entre 5 y 10 mg semanales en pulso (no a diario), con o sin pausas periódicas, buscando preservar mTORC2 y minimizar efectos metabólicos. Ningún médico responsable la prescribe sin monitorización.
¿Qué es mejor para la mitofagia cerebral, ejercicio o rapamicina?
En humanos sanos, hoy: el ejercicio. Activa AMPK→ULK1 + PGC-1α en paralelo, eleva lactato y BDNF cerebrales, tiene RCTs cardiovasculares y cognitivos con décadas de datos, no requiere prescripción y no penetra peor que un fármaco que resulta indetectable en LCR. La rapamicina sigue siendo experimental.
¿Está aprobada la rapamicina para el Alzheimer en España?
No. La AEMPS y la EMA mantienen las indicaciones autorizadas en prevención de rechazo de trasplantes y algunas indicaciones oncológicas. Para Alzheimer, longevidad o cognición, la prescripción es off-label y depende del criterio clínico individual del prescriptor, que asume la responsabilidad.
Este artículo es contenido editorial de KRECE. No sustituye al criterio médico individualizado. Las decisiones sobre uso de rapamicina, sirolimus, everolimus o cualquier otro fármaco geroprotector corresponden al médico tratante, que conoce la historia clínica completa y la legislación aplicable. La rapamicina para indicaciones de longevidad o neurodegeneración es prescripción off-label en España, la UE y EE. UU. y no debe iniciarse sin supervisión clínica. Los datos cuantitativos citados provienen de artículos verificados con la fuente primaria; la interpretación editorial es de KRECE y puede actualizarse con nuevas evidencias.
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12(1):9-14. DOI.
- Laker RC, Drake JC, Wilson RJ, et al. AMPK phosphorylation of ULK1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun. 2017;8:548. PMID 28916822.
- Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013;20(1):31-42. PMID 22743996.
- Kerr JS, Adriaanse BA, Greig NH, et al. Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci. 2017;40(3):151-166. PMID 28190529.
- Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature. 2015;521(7553):525-528. PMID 25896323.
- Lamming DW, Ye L, Katajisto P, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012;335(6076):1638-1643. PMID 22461615.
- Halloran J, Hussong SA, Burbank R, et al. Chronic inhibition of mammalian target of rapamycin by rapamycin modulates cognitive and non-cognitive components of behavior. Neuroscience. 2012;223:102-113. PMID 22750207.
- Gonzales MM, Garbarino VR, Kautz TF, et al. Rapamycin treatment for Alzheimer’s disease and related dementias: a pilot phase 1 clinical trial. Commun Med (Lond). 2025;5:189. DOI. PMID 40394335. PMCID PMC12092812.
- Wang H, Fu J, Xu X, Yang Z, Zhang T. Rapamycin activates mitophagy and alleviates cognitive and synaptic plasticity deficits in a mouse model of Alzheimer’s disease. J Gerontol A Biol Sci Med Sci. 2021;76(10):1707-1713. DOI. PMID 34003967.
- Reid JJ, Linden MA, Peelor FF, Miller RA, Hamilton KL, Miller BF. Brain protein synthesis rates in the UM-HET3 mouse following treatment with rapamycin or rapamycin with metformin. J Gerontol A Biol Sci Med Sci. 2020;75(1):40-49. DOI. PMID 30864661.
- Drake JC, Wilson RJ, Laker RC, et al. Mitochondria-localized AMPK responds to local energetics and contributes to exercise and energetic stress-induced mitophagy. Proc Natl Acad Sci USA. 2021;118(37):e2025932118. PMID 34504024.
- Mito T, Vincent AE, Faitg J, et al. Mosaic dysfunction of mitophagy in mitochondrial muscle disease. Cell Metab. 2022;34(2):197-208.e5. DOI. PMID 35030325.
- Kaeberlein M, Galvan V. Rapamycin and Alzheimer’s disease: time for a clinical trial? Sci Transl Med. 2019;11(476):eaar4289. DOI. PMID 30674654.
- Carosi JM, Sargeant TJ. Rapamycin and Alzheimer disease: a double-edged sword? Autophagy. 2019;15(8):1460-1462. DOI. PMID 31066320.
- UT Health San Antonio. Large rapamycin clinical trial launches at UT Health San Antonio. Nota de prensa, 25 de marzo de 2026. Ensayo multi-fase financiado por el NIA en mayores (n=84). news.uthscsa.edu.
- Mannick JB, Del Giudice G, Lattanzi M, et al. mTOR inhibition improves immune function in the elderly. Sci Transl Med. 2014;6(268):268ra179. DOI. PMID 25540326.
- Mannick JB, Morris M, Hockey HP, et al. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci Transl Med. 2018;10(449):eaaq1564. PMID 29997249.
- Ryu D, Mouchiroud L, Andreux PA, et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med. 2016;22(8):879-888. PMID 27400265.
- Eisenberg T, Abdellatif M, Schroeder S, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. 2016;22(12):1428-1438. PMID 27841876.
- Wirth M, Benson G, Schwarz C, et al. The effect of spermidine on memory performance in older adults at risk for dementia: a randomized controlled trial (ensayo piloto del programa SmartAge). Cortex. 2018;109:181-188. DOI. PMID 30388439.
- Schönfeld P, Reiser G. Why does brain metabolism not favor burning of fatty acids to provide energy? J Cereb Blood Flow Metab. 2013;33(10):1493-1499. PMID 23921897.
- López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: an expanding universe. Cell. 2023;186(2):243-278. DOI. PMID 36599349.