
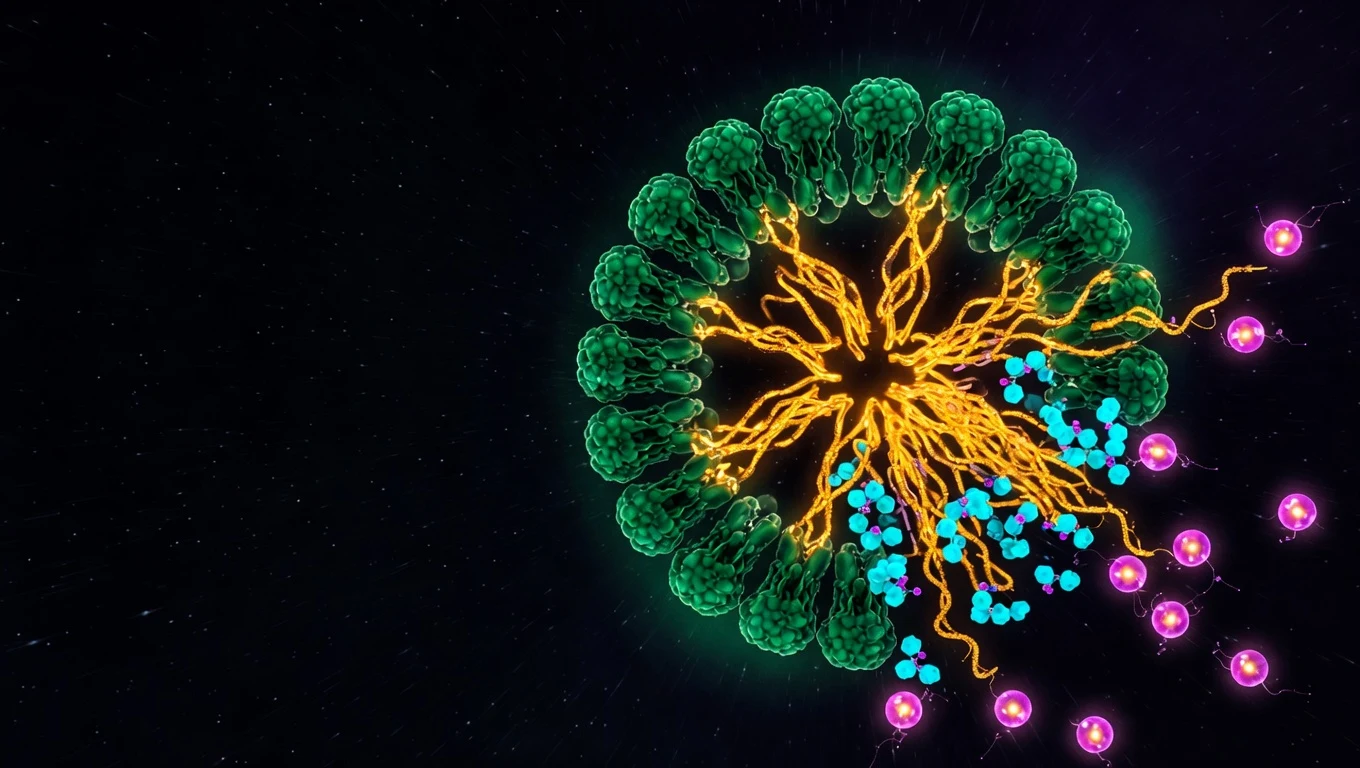
NLRP3 es el sensor de inmunidad innata que detecta daño celular estéril (DAMPs) y ensambla un complejo multiproteico — el inflamasoma — que activa caspasa-1 para producir IL-1β, IL-18 y piroptosis. Es el motor molecular del inflammaging.
El término «inflamasoma» fue acuñado por Jürg Tschopp en 2002; NLRP3 (NOD-, LRR- y Pyrin domain-containing protein 3, antes NALP3 o criopirina) es el más estudiado por su rol en inflamación estéril. A diferencia de los sensores de patógenos, NLRP3 detecta señales de peligro propias: cristales (urato en gota, colesterol en aterosclerosis, β-amiloide en Alzheimer), ATP extracelular, ADN mitocondrial, ácidos grasos saturados. Eicke Latz (Bonn) documentó su rol central en el inflammaging — la inflamación estéril crónica de bajo grado del envejecimiento. NLRP3 procesa el IL-1β que forma parte del SASP de las células senescentes, conectando senescencia e inflamación en un bucle. La validación clínica llegó con CANTOS (canakinumab, anti-IL-1β) que redujo eventos cardiovasculares — confirmando la hipótesis inflamatoria de la aterosclerosis. El interés de big pharma culminó con la compra de Ventyx por Eli Lilly (~1.200 millones, enero 2026) por su plataforma de inhibidores NLRP3. Esta entrada KRECE define NLRP3 molecularmente, articula el ensamblaje en dos señales, contrasta vía canónica y no canónica, mapea las enfermedades NLRP3-mediadas, y documenta el pipeline 2026 de inhibidores.
El sensor que convierte el daño celular en inflamación. NLRP3 es el detector de «peligro estéril» del cuerpo — y cuando se activa crónicamente con la edad, se convierte en uno de los motores moleculares del envejecimiento.
NLRP3 es un sensor de inmunidad innata que forma el núcleo del inflamasoma — un complejo multiproteico citosólico que, al detectar señales de daño (DAMPs), ensambla una plataforma de activación de caspasa-1. Caspasa-1 procesa pro-IL-1β y pro-IL-18 a sus formas activas y escinde gasdermina D para inducir piroptosis — muerte celular inflamatoria. Acuñado el concepto por Jürg Tschopp en 2002, NLRP3 es el inflamasoma más estudiado por su papel en inflamación estéril.
La relevancia editorial es enorme: NLRP3 es el nexo molecular entre estrés metabólico/celular e inflamación crónica. Procesa el IL-1β que forma parte del SASP de las células senescentes — conectando senescencia e inflammaging en un bucle de retroalimentación. Es el mecanismo detrás de la gota (cristales de urato), la aterosclerosis (cristales de colesterol, validado por el ensayo CANTOS) y la neuroinflamación del Alzheimer (β-amiloide). El campo terapéutico 2026 está en ebullición: la única terapia antiinflamatoria aprobada FDA para enfermedad coronaria es la colchicina (2023); ningún inhibidor directo de NLRP3 está aprobado aún, pero el pipeline es intenso — y Eli Lilly compró Ventyx por ~1.200 millones (enero 2026) por su plataforma de inhibidores NLRP3, con VTX3232 (penetrante en SNC) en Phase 2 para Parkinson. Esta entrada KRECE define NLRP3 molecularmente, articula el ensamblaje en dos señales, mapea las enfermedades NLRP3-mediadas, y documenta el pipeline completo.
¿Qué es NLRP3?
Definición bioquímica
NLRP3 (NOD-, LRR- and Pyrin domain-containing protein 3) — también conocido históricamente como NALP3 o criopirina — es un sensor de inmunidad innata de la familia NLR (NOD-like receptors). Su función es formar el núcleo del inflamasoma NLRP3: un complejo multiproteico citosólico que actúa como plataforma de activación de caspasa-1, desencadenando la respuesta inflamatoria.
A diferencia de los sensores que detectan patógenos específicos, NLRP3 es un sensor de «peligro estéril» — responde a una amplia gama de señales de daño celular (DAMPs, Damage-Associated Molecular Patterns) sin necesidad de infección. Esta amplitud de detección es lo que lo convierte en el inflamasoma más estudiado y más relevante para enfermedades inflamatorias crónicas no infecciosas.
Estructura de dominios de NLRP3
La proteína NLRP3 tiene tres dominios funcionales que explican su mecanismo:
| Dominio | Posición | Función |
|---|---|---|
| PYD (Pyrin Domain) | N-terminal | Reclutamiento de la proteína adaptadora ASC vía interacción PYD-PYD |
| NACHT (con actividad ATPasa) | Central | Oligomerización — el motor del ensamblaje. Requiere unión e hidrólisis de ATP |
| LRR (Leucine-Rich Repeat) | C-terminal | Autoinhibición en estado de reposo y sensado/modulación |
En estado de reposo, el dominio LRR mantiene a NLRP3 en una conformación autoinhibida. La activación libera esta autoinhibición y permite la oligomerización vía NACHT.
Componentes del inflamasoma ensamblado
El inflamasoma NLRP3 funcional es un complejo de tres componentes:
- NLRP3: el sensor. Detecta la señal de activación y oligomeriza.
- ASC (Apoptosis-associated Speck-like protein containing a CARD): la proteína adaptadora. Tiene dominios PYD (une NLRP3) y CARD (une caspasa-1). Polimeriza formando el «ASC speck» — una estructura micrométrica visible al microscopio que es marcador de activación.
- Pro-caspasa-1: la enzima efectora inactiva, reclutada al complejo vía interacción CARD-CARD con ASC.
Función: una plataforma de activación de caspasa-1
El inflamasoma NLRP3 no actúa directamente sobre las citoquinas — actúa concentrando y activando caspasa-1. Cuando NLRP3 oligomeriza y recluta ASC y pro-caspasa-1, la proximidad física induce autoproteolisis de pro-caspasa-1 a caspasa-1 activa. Esta caspasa-1 activa es la que:
- Procesa pro-IL-1β a IL-1β maduro (activo)
- Procesa pro-IL-18 a IL-18 maduro (activo)
- Escinde gasdermina D para inducir piroptosis
Historia: Tschopp y el descubrimiento del inflamasoma
El concepto de «inflamasoma» fue acuñado por Jürg Tschopp y colaboradores (Universidad de Lausana) en 2002, al describir el complejo multiproteico que activa caspasas inflamatorias. NLRP3 emergió rápidamente como el inflamasoma de mayor relevancia clínica por dos razones:
- Amplitud de activadores: responde a la gama más diversa de estímulos (cristales, ATP, toxinas, ROS, agregados proteicos).
- Enfermedades autoinflamatorias: las mutaciones gain-of-function en NLRP3 causan los síndromes periódicos asociados a criopirina (CAPS) — validación humana directa de su rol patogénico.
Por qué importa
NLRP3 es el nexo molecular entre estrés celular/metabólico e inflamación. Su importancia editorial deriva de su centralidad en múltiples ejes:
- Inflammaging: NLRP3 se activa crónicamente con la edad por acumulación de DAMPs — es uno de los motores moleculares de la inflamación estéril crónica del envejecimiento.
- Senescencia: procesa el IL-1β que forma parte del SASP — conexión directa con biología de la senescencia.
- Enfermedades crónicas: gota, aterosclerosis, Alzheimer, diabetes tipo 2, enfermedad renal crónica — todas tienen componente NLRP3.
- Diana terapéutica: el interés farmacológico es intenso, culminando en la compra de Ventyx por Eli Lilly en enero 2026.
La centralidad de NLRP3 en el marco de gerociencia (gerociencia) es que conecta el hallmark de «inflamación crónica» con casi todos los demás: detecta los productos del daño genómico (mtDNA citosólico), de la disfunción mitocondrial (ROS), de la senescencia (DAMPs del SASP), y del desequilibrio metabólico (cristales de colesterol, ácidos grasos). Es, en cierto sentido, el «detector de humo» molecular del envejecimiento — lo cual lo convierte en diana atractiva pero también peligrosa: inhibirlo crónicamente significa silenciar un sistema de alarma con funciones defensivas legítimas.
Ensamblaje del inflamasoma NLRP3: las dos señales
El modelo de dos señales
La activación del inflamasoma NLRP3 requiere dos señales secuenciales — un mecanismo de seguridad que evita activación inapropiada. Esta arquitectura de dos pasos es central para entender tanto su regulación como las estrategias de inhibición.
Señal 1: Priming (cebado)
La primera señal prepara el sistema. Un estímulo inflamatorio inicial activa la transcripción de los componentes:
- Estímulos típicos: ligandos de TLR (como LPS bacteriano), TNF-α, IL-1β — señales que activan NF-κB.
- Efecto transcripcional: NF-κB induce la transcripción de NLRP3 y de pro-IL-1β. Los niveles basales de ambos son insuficientes — el priming los eleva a niveles funcionales.
- Efecto no transcripcional (priming rápido): también hay modificaciones post-traduccionales de NLRP3 (desubiquitinación, fosforilación) que lo «licencian» para activación.
El priming explica por qué la inflamación preexistente amplifica la respuesta NLRP3 — un círculo vicioso relevante en inflammaging, donde el tono inflamatorio basal elevado ceba continuamente el sistema.
Señal 2: Activación (ensamblaje)
La segunda señal desencadena el ensamblaje del complejo. Múltiples activadores diversos convergen en eventos celulares comunes (desarrollados en sección 04):
- Eflujo de potasio (K⁺ efflux): el evento más universal. La caída de K⁺ intracelular es disparador común a casi todos los activadores.
- Disfunción mitocondrial: liberación de ROS mitocondrial y ADN mitocondrial al citosol.
- Desestabilización lisosomal: cristales fagocitados rompen el lisosoma, liberando catepsina B.
El ensamblaje físico paso a paso
Una vez recibida la señal 2, el ensamblaje procede en cascada:
- NLRP3 se libera de autoinhibición y oligomeriza vía dominio NACHT (dependiente de ATP).
- NLRP3 recluta ASC vía interacción PYD-PYD.
- ASC polimeriza formando filamentos que condensan en el «ASC speck» — una estructura micrométrica única por célula, visible al microscopio y marcador definitorio de activación del inflamasoma.
- ASC recluta pro-caspasa-1 vía interacción CARD-CARD.
- La proximidad induce autoproteolisis de pro-caspasa-1 a caspasa-1 activa.
NEK7: el licenciador del ensamblaje
Un descubrimiento clave de la última década es el papel de NEK7 (NIMA-related kinase 7). NEK7 es una kinasa que se une a NLRP3 y es esencial para su oligomerización — actúa como un «licenciador» del ensamblaje. La interacción NLRP3-NEK7 es necesaria para la activación del inflamasoma y representa una diana terapéutica de interés (interferir esa interacción bloquea NLRP3 sin afectar otros inflamasomas).
Vía canónica vs no canónica
El ensamblaje descrito arriba es la vía canónica (dependiente de caspasa-1). Existe también una vía no canónica:
| Característica | Vía canónica | Vía no canónica |
|---|---|---|
| Caspasa efectora | Caspasa-1 | Caspasa-4, -5 (humano), caspasa-11 (ratón) |
| Sensor del estímulo | NLRP3 + ASC | Las caspasas reconocen LPS citosólico directamente |
| Procesa IL-1β/IL-18 | Sí (directamente) | Indirectamente (vía activación NLRP3 secundaria) |
| Induce piroptosis | Sí (gasdermina D) | Sí (gasdermina D + K⁺ efflux que activa NLRP3 canónico) |
La vía no canónica detecta LPS intracelular (de bacterias Gram-negativas que escaparon al citosol) sin necesidad de receptor de superficie. Ambas vías convergen en gasderdmina D y piroptosis.
Productos del inflamasoma: IL-1β, IL-18 y piroptosis
La activación del inflamasoma NLRP3 genera tres productos efectores principales, todos mediados por caspasa-1 (vía canónica).
IL-1β: la citoquina pro-inflamatoria maestra
IL-1β (interleuquina-1 beta) es el producto más relevante clínicamente del inflamasoma:
- Procesamiento: se sintetiza como pro-IL-1β inactivo (durante el priming). Caspasa-1 lo escinde a IL-1β maduro activo.
- Secreción no convencional: IL-1β no tiene péptido señal clásico — se libera por poros de gasdermina D o durante piroptosis, no por la vía secretora clásica.
- Efectos: reclutamiento de neutrófilos, inducción de fiebre (pirógeno endógeno), activación endotelial, amplificación de la cascada inflamatoria (induce más IL-6, TNF-α).
- Bucle de retroalimentación: IL-1β es ella misma una señal de priming — activa NF-κB en células vecinas, amplificando la respuesta. Esto explica la naturaleza autoamplificada de la inflamación NLRP3.
- Diana terapéutica: canakinumab (anti-IL-1β), anakinra (antagonista del receptor IL-1), rilonacept (trampa de IL-1) bloquean este eje downstream.
IL-18: el activador de la respuesta Th1/NK
IL-18 (interleuquina-18) es el segundo producto citoquínico:
- Procesamiento: similar a IL-1β, se procesa de pro-IL-18 por caspasa-1. A diferencia de pro-IL-1β, pro-IL-18 se expresa constitutivamente (no requiere priming intenso).
- Efectos: induce producción de IFN-γ, activa células NK y respuesta Th1. Más relevante en inmunidad antiviral/antitumoral.
- Relevancia en envejecimiento: IL-18 elevado es marcador de inflammaging y se asocia a sarcopenia, fragilidad y mortalidad.
Piroptosis: la muerte celular inflamatoria
El tercer producto no es una molécula sino un tipo de muerte celular: la piroptosis, mediada por gasdermina D.
- Mecanismo: caspasa-1 escinde gasdermina D (GSDMD). El fragmento N-terminal liberado se inserta en la membrana plasmática formando poros.
- Consecuencias de los poros:
- Liberación de IL-1β e IL-18 maduros al espacio extracelular.
- Eflujo de K⁺ e influjo de agua → hinchazón osmótica → lisis celular.
- Liberación de DAMPs adicionales (HMGB1, ATP, mtDNA) — que activan NLRP3 en células vecinas.
- Diferencia con apoptosis: la apoptosis es muerte «silenciosa» no inflamatoria (membrana intacta, cuerpos apoptóticos). La piroptosis es lítica e inflamatoria — rompe la membrana y vierte contenido pro-inflamatorio.
- Función biológica: defensa antimicrobiana (elimina nicho replicativo de patógenos intracelulares) pero patológica cuando es crónica o excesiva.
La familia gasdermina
Gasdermina D es la principal pero no la única ejecutora de piroptosis:
- GSDMD (gasdermina D): la canónica, escindida por caspasa-1/-4/-5/-11.
- GSDME (gasdermina E): escindida por caspasa-3 — actúa como switch entre apoptosis y piroptosis en células con alta expresión GSDME (relevante en cáncer). Los linfocitos Th17 humanos usan poros de GSDME para liberar IL-1α tras activación NLRP3.
La lógica de por qué NLRP3 es tan dañino crónicamente: cada activación no solo libera citoquinas, sino que mata la célula de forma inflamatoria (piroptosis) y libera DAMPs que activan NLRP3 en células vecinas. Es un mecanismo autoamplificado — igual que el SASP de la senescencia. De hecho, ambos sistemas se entrelazan: el SASP libera DAMPs que activan NLRP3, y NLRP3 produce el IL-1β del SASP. En el envejecimiento, estos dos bucles se refuerzan mutuamente, generando la inflamación estéril crónica que define al inflammaging.
Activadores y mecanismos de activación del inflamasoma NLRP3
El enigma de la diversidad de activadores
Una de las características más notables de NLRP3 es la enorme diversidad de estímulos que lo activan — cristales, toxinas, ATP, agregados proteicos, virus, hongos. Esto planteó un enigma: ¿cómo puede un solo sensor detectar tantos estímulos químicamente dispares? La respuesta: NLRP3 no detecta los activadores directamente, sino que detecta eventos celulares comunes que todos ellos provocan.
Eflujo de potasio: el denominador común
El eflujo de K⁺ (caída del potasio intracelular) es el evento más universal en la activación NLRP3:
- ATP extracelular: actúa sobre el receptor P2X7, abriendo un canal que permite eflujo masivo de K⁺. El ATP extracelular es DAMP clásico liberado por células dañadas/muertas.
- Toxinas formadoras de poros: nigericina (herramienta experimental), toxinas bacterianas — crean poros que permiten eflujo de K⁺.
- Mecanismo: la caída de K⁺ citosólico por debajo de un umbral crítico es señal directa para ensamblaje NLRP3.
Disfunción mitocondrial y ROS
Las mitocondrias dañadas son fuente central de activación NLRP3:
- ROS mitocondrial (mtROS): las especies reactivas de oxígeno de mitocondrias disfuncionales activan NLRP3.
- ADN mitocondrial (mtDNA) citosólico: cuando mitocondrias dañadas liberan su ADN al citosol, este actúa como DAMP que se une a NLRP3 y activa el ensamblaje. También activa cGAS-STING en paralelo.
- Cardiolipina: este lípido mitocondrial, normalmente interno, se externaliza en disfunción y une directamente NLRP3.
- Relevancia en envejecimiento: la disfunción mitocondrial acumulada con la edad es fuente crónica de activación NLRP3 — mecanismo del inflammaging.
Desestabilización lisosomal: el mecanismo de los cristales
Los activadores cristalinos actúan vía un mecanismo distintivo:
- Fagocitosis: la célula (típicamente macrófago) fagocita el cristal (urato, colesterol, sílice, asbesto, β-amiloide).
- Ruptura lisosomal: el cristal es demasiado grande/duro para ser digerido y rompe la membrana lisosomal.
- Liberación de catepsina B: esta proteasa lisosomal se libera al citosol y contribuye a activar NLRP3.
- Relevancia clínica directa: explica gota (cristales de urato), aterosclerosis (cristales de colesterol), silicosis (sílice), Alzheimer (fibrillas β-amiloide).
Tabla de activadores por categoría
| Categoría | Activadores | Enfermedad asociada |
|---|---|---|
| Cristales / partículas | Urato monosódico (MSU), colesterol, sílice, asbesto, alum, β-amiloide | Gota, aterosclerosis, silicosis, Alzheimer |
| DAMPs endógenos | ATP extracelular, mtDNA, HMGB1, ácido úrico, S100 | Inflammaging, senescencia (SASP) |
| Metabólicos | Ácidos grasos saturados (palmitato), ceramidas, glucosa alta, islet amyloid polypeptide (IAPP) | Diabetes tipo 2, NAFLD, obesidad |
| Patógenos / PAMPs | Toxinas formadoras de poros, ARN viral, hifas fúngicas | Infecciones |
| Ambientales | UV, partículas de polución, nanopartículas | Daño cutáneo, inflamación pulmonar |
Reguladores negativos y el eje NAD⁺
NLRP3 tiene reguladores negativos cuya pérdida con la edad contribuye a su hiperactivación:
- Autofagia/mitofagia: la eliminación de mitocondrias dañadas (fuente de mtROS/mtDNA) reduce activación NLRP3. El declive de autofagia con la edad aumenta la activación.
- Sirtuinas y NAD⁺: SIRT2 desacetila NLRP3 reduciendo su activación. El declive de NAD⁺ con la edad (parcialmente por consumo vía CD38) reduce actividad sirtuina y desinhibe NLRP3. Conexión con el eje NAD⁺ cubierto en sirtuinas.
- Cetonas (BHB): el β-hidroxibutirato es inhibidor endógeno de NLRP3 — mecanismo parcial de los beneficios antiinflamatorios del ayuno y la restricción calórica.
Dato accionable: el β-hidroxibutirato (BHB), producido durante ayuno, ejercicio prolongado o dieta cetogénica, es un inhibidor endógeno directo de NLRP3. Esto proporciona un mecanismo molecular concreto para los efectos antiinflamatorios documentados del ayuno intermitente: al elevar BHB, se reduce la activación del inflamasoma. Es una de las pocas palancas no farmacológicas con mecanismo claro sobre NLRP3.
NLRP3 como motor del inflammaging y la senescencia
NLRP3 y el concepto de inflammaging
El inflammaging — término acuñado por Claudio Franceschi — describe la inflamación estéril crónica de bajo grado que caracteriza al envejecimiento. NLRP3 es uno de sus motores moleculares centrales, como documentó Eicke Latz (Bonn) en su revisión de 2018 (Semin Immunol):
- Acumulación de DAMPs con la edad: mtDNA de mitocondrias dañadas, cristales de colesterol, agregados proteicos, ácidos grasos libres — todos se acumulan con la edad y activan NLRP3 crónicamente.
- Metaflammation: Latz acuñó el concepto de que el exceso calórico y el estilo de vida moderno agravan la activación NLRP3 — «metaflammation» (inflamación metabólica).
- Producción crónica de IL-1β/IL-18: el resultado es inflamación sistémica de bajo grado persistente.
- Validación animal: ratones NLRP3 knockout muestran healthspan extendido, menor declive cognitivo, mejor función metabólica y ósea con la edad — demostrando rol causal de NLRP3 en el envejecimiento inflamatorio.
El loop NLRP3-senescencia-SASP
La conexión más relevante editorialmente: NLRP3 y la senescencia celular forman un bucle de retroalimentación que es central en el envejecimiento. El SASP (fenotipo secretor asociado a senescencia) y NLRP3 se entrelazan:
- NLRP3 produce el IL-1β del SASP: el IL-1β es componente nuclear del SASP, y es procesado precisamente por el inflamasoma NLRP3 (vía caspasa-1). Sin NLRP3 funcional, el componente IL-1β del SASP se reduce.
- El SASP activa NLRP3 en células vecinas: los DAMPs del SASP (HMGB1, mtDNA, ATP) activan NLRP3 en células sanas adyacentes.
- cGAS-STING conecta ambos: el ADN citosólico de células senescentes activa cGAS-STING, que a su vez puede activar NLRP3.
- Resultado — espiral inflamatoria: senescencia → SASP → activación NLRP3 en células vecinas → más IL-1β → más senescencia paracrina → más SASP. Bucle autoamplificado del inflammaging.
Esta conexión es por qué KRECE trata NLRP3 y SASP como entradas complementarias: son las dos caras de la inflamación del envejecimiento — el inflamasoma (sensor/productor) y el secretoma senescente (fuente de señales).
NLRP3 y los hallmarks del envejecimiento
NLRP3 se conecta con múltiples hallmarks of aging:
- Inflamación crónica (hallmark añadido 2023): NLRP3 es uno de sus motores principales.
- Disfunción mitocondrial: las mitocondrias dañadas activan NLRP3 (mtROS, mtDNA).
- Senescencia celular: NLRP3 procesa el IL-1β del SASP.
- Comunicación intercelular alterada: IL-1β e IL-18 son señales paracrinas aberrantes.
- Desregulación del sensado de nutrientes: el exceso metabólico (palmitato, glucosa) activa NLRP3 — conexión con metaflammation.
NLRP3 e inmunosenescencia
Hay una paradoja interesante en la relación NLRP3-envejecimiento inmune: mientras NLRP3 está crónicamente hiperactivado (inflammaging), la inmunidad adaptativa declina (inmunosenescencia). El resultado es un sistema inmune que está simultáneamente hiperinflamado (innato) y debilitado (adaptativo) — mala inflamación crónica estéril junto a mala respuesta a patógenos reales y vacunas. Esta disociación es uno de los rasgos centrales del envejecimiento inmunológico.
NLRP3 en enfermedades: gota, cardiovascular, Alzheimer y más
Gota: el caso paradigmático
La gota es el ejemplo más claro y directo de enfermedad NLRP3-mediada:
- Mecanismo: los cristales de urato monosódico (MSU) que precipitan en articulaciones son fagocitados por macrófagos → rompen el lisosoma → activan NLRP3 → IL-1β → inflamación articular aguda intensa (el ataque de gota).
- Validación terapéutica: los anti-IL-1 (canakinumab, anakinra) son altamente eficaces en gota refractaria — confirmando que el daño es IL-1β-mediado.
- Colchicina: el tratamiento clásico de la gota inhibe el ensamblaje NLRP3 (vía microtúbulos).
Cardiovascular: la hipótesis inflamatoria y CANTOS
La aterosclerosis tiene un componente NLRP3 central, y su validación clínica es uno de los hitos de la cardiología moderna:
- Mecanismo: los cristales de colesterol en la placa aterosclerótica activan NLRP3 en macrófagos → IL-1β → inflamación vascular → progresión e inestabilidad de placa.
- CANTOS (2017): el ensayo Canakinumab Anti-Inflammatory Thrombosis Outcome Study (Ridker, NCT01327846) incluyó más de 10.000 pacientes post-infarto con hsCRP elevada. Canakinumab (anti-IL-1β) redujo eventos cardiovasculares mayores ~15% independientemente de los lípidos — la primera prueba directa de la «hipótesis inflamatoria» de la aterosclerosis.
- El caveat de CANTOS: canakinumab también aumentó el riesgo de infección fatal y NO obtuvo aprobación FDA como terapia cardiovascular (coste/beneficio desfavorable a esa escala). Pero estableció el proof-of-concept.
- CIRT (methotrexate) FALLÓ: el ensayo con metotrexato no redujo eventos CV — y notablemente, no bajó IL-1β/IL-6/CRP. Esto refuerza la especificidad de la vía IL-1/NLRP3 (no cualquier antiinflamatorio sirve).
- Colchicina: la validación práctica: LoDoCo2 y COLCOT demostraron que colchicina 0,5 mg/día reduce eventos CV (~23% en LoDoCo2, HR 0,77). La colchicina (Lodoco) fue aprobada por FDA en 2023 — el primer (y a 2026 único) antiinflamatorio aprobado para reducción de riesgo CV en enfermedad coronaria establecida. Barata, antigua, accesible. Cobertura del eje en inflammaging.
Alzheimer y neuroinflamación
NLRP3 tiene rol creciente en la patología de Alzheimer:
- Activación por β-amiloide: las fibrillas de β-amiloide son fagocitadas por microglía → ruptura lisosomal → activación NLRP3 → neuroinflamación.
- ASC specks siembran amiloide: un hallazgo notable — los ASC specks liberados por microglía piroptótica actúan como semilla de agregación de β-amiloide (cross-seeding). NLRP3 no solo responde al amiloide sino que acelera su agregación.
- Validación animal: ratones NLRP3 KO o caspasa-1 KO en modelos de Alzheimer muestran menos placa y mejor cognición.
- Pipeline: inhibidores NLRP3 brain-penetrant (como VTX3232) son candidatos para Alzheimer. Cobertura en cinco moléculas para prevenir Alzheimer.
Parkinson y neuroinflamación
Parkinson es donde el pipeline NLRP3 está más avanzado clínicamente:
- Mecanismo: α-sinucleína agregada activa NLRP3 en microglía → neuroinflamación → daño de neuronas dopaminérgicas en sustancia nigra.
- VTX3232 (Ventyx/Eli Lilly): inhibidor NLRP3 penetrante en SNC, en Phase 2 para Parkinson precoz con datos prometedores (ver sección 07). Conexión con neuroinflamación y BDNF.
Enfermedades metabólicas
- Diabetes tipo 2: ácidos grasos saturados (palmitato), ceramidas, IAPP (islet amyloid polypeptide) e hiperglucemia activan NLRP3 en islotes e hígado → resistencia insulínica y disfunción de células beta.
- NAFLD/NASH: NLRP3 contribuye a la progresión de hígado graso a esteatohepatitis y fibrosis.
- Obesidad: el tejido adiposo de obesos tiene NLRP3 hiperactivado, conectando inflamación con disfunción metabólica.
CAPS: la validación humana directa
Los síndromes periódicos asociados a criopirina (CAPS) son enfermedades autoinflamatorias raras causadas por mutaciones gain-of-function en el gen NLRP3. Son la prueba humana más directa del rol patogénico de NLRP3:
- Las mutaciones hacen a NLRP3 hiperactivo — se ensambla sin necesidad de señal 2 adecuada.
- Resultado: fiebre recurrente, urticaria, artralgia, riesgo de amiloidosis.
- Respuesta dramática a anti-IL-1: canakinumab y anakinra revierten los síntomas — confirmando que el daño es IL-1β-mediado vía NLRP3.
Microbiota e intestino
NLRP3 tiene rol complejo en el intestino: por un lado mantiene homeostasis de barrera, por otro su activación excesiva contribuye a enfermedad inflamatoria intestinal. La disbiosis altera la activación NLRP3. Cobertura del eje en eje microbiota-autoinmunidad.
Inhibidores de NLRP3 en 2026: del MCC950 a la compra de Ventyx
Tres estrategias de inhibición
La modulación farmacológica de la vía NLRP3 opera en tres niveles distintos:
| Estrategia | Diana | Ventaja / Desventaja |
|---|---|---|
| Inhibición directa de NLRP3 | El sensor NLRP3 mismo | Más selectivo (no afecta otros inflamasomas) / clase nueva, sin aprobaciones aún |
| Inhibición downstream (anti-IL-1) | IL-1β o su receptor | Validado clínicamente (CANTOS, CAPS) / bloqueo global de IL-1, riesgo infección |
| Inhibición indirecta | Ensamblaje (microtúbulos) | Barato, disponible (colchicina) / menos selectivo, efectos GI |
Inhibidores directos de NLRP3: la clase emergente
MCC950 (CRID3): el prototipo de investigación
- Mecanismo: bloquea NLRP3 manteniiéndolo en conformación inactiva (se une al dominio NACHT, impide hidrólisis de ATP necesaria para oligomerización).
- Potencia: el inhibidor directo de NLRP3 más estudiado y la herramienta de referencia preclínica.
- Limitación: NO avanzó clínicamente por hepatotoxicidad en ensayos tempranos (originalmente desarrollado por Pfizer como CRID3). Sigue siendo la herramienta experimental estándar.
VTX3232 y VTX2735 (Ventyx Biosciences → Eli Lilly)
El programa NLRP3 más relevante comercialmente a 2026 — y el que motivó la mayor operación del año en la clase:
- VTX3232: inhibidor NLRP3 penetrante en SNC (brain-penetrant). Este es su diferencial clave — permite atacar neuroinflamación central.
- Phase 2 en Parkinson precoz (presentado en MDS 2025, Honolulu): bien tolerado, sin eventos adversos relacionados con el fármaco entre los pacientes early PD; mejora en síntomas motores y no motores (escala MDS-UPDRS); reducciones medibles de IL-1β e IL-18 en SNC.
- Phase 2 en obesidad y factores de riesgo cardiovascular (NCT06771115): reducciones significativas en marcadores de riesgo CV.
- Planes para Phase 2 placebo-controlado en Parkinson y potencialmente Alzheimer.
- VTX2735: inhibidor NLRP3 periférico (no penetra SNC), en Phase 2 para pericarditis recurrente (NCT06836232).
- La compra de Eli Lilly: el 7 de enero de 2026, Eli Lilly anunció la adquisición de Ventyx Biosciences por ~1.200 millones de dólares (14 dólares por acción, prima del 62% sobre el precio medio ponderado de 30 días). Fue la primera operación de M&A de Lilly en 2026. Sanofi tenía un derecho de primera negociación (tras una inversión estratégica previa de 27 millones) pero Lilly se adelantó.
- Lectura del mercado: la operación valida el interés de big pharma en la clase NLRP3 para enfermedades neuroinflamatorias, cardiometabólicas e inflamatorias. Como apuntó el analista Myles Minter (William Blair), «lee positivamente para otros inhibidores NLRP3 en desarrollo».
Otros inhibidores directos en pipeline
- Dapansutrile (OLT1177, Olatec): inhibidor NLRP3 oral con buen perfil de seguridad. Phase 2 en gota, insuficiencia cardíaca, síndrome de Schnitzler.
- NodThera (NT-0796, NT-0249): inhibidores NLRP3 brain-penetrant. Phase 1/2 en condiciones cardiometabólicas y neuroinflamatorias.
- Selnoflast (Roche/IFM Therapeutics), ZYIL1 (Zydus), y otros en fases tempranas.
- Disulfiram: fármaco aprobado FDA para alcoholismo, descubierto que bloquea la formación del poro de gasdermina D — candidato de repurposing para condiciones inflamatorias crónicas.
Inhibidores downstream (anti-IL-1): ya aprobados
| Fármaco | Mecanismo | Estatus 2026 |
|---|---|---|
| Canakinumab (Ilaris) | Anticuerpo monoclonal anti-IL-1β | Aprobado FDA para CAPS, gota refractaria, Still. CANTOS positivo pero NO aprobado para CV. |
| Anakinra (Kineret) | Antagonista del receptor IL-1 | Aprobado FDA para AR, CAPS. Vida media corta. |
| Rilonacept (Arcalyst) | Trampa de IL-1 (receptor soluble) | Aprobado FDA para CAPS, pericarditis recurrente. |
Inhibición indirecta: la colchicina
La colchicina merece atención especial porque es la única terapia antiinflamatoria con aprobación para enfermedad cardiovascular establecida:
- Mecanismo: inhibe la polimerización de microtúbulos, interfiriendo con el ensamblaje del inflamasoma NLRP3 (que requiere transporte microtubular). También reduce adhesión/migración de neutrófilos.
- Historia: fármaco antiquísimo (extracto de Colchicum autumnale), tratamiento clásico de la gota.
- LoDoCo2 y COLCOT: ensayos que demostraron reducción de eventos CV (~23% en LoDoCo2, HR 0,77; 95% CI 0,61-0,96) con colchicina 0,5 mg/día en enfermedad coronaria.
- Aprobación FDA 2023: la colchicina low-dose (marca Lodoco) fue aprobada por la FDA en 2023 para reducción de riesgo cardiovascular — el primer y único antiinflamatorio aprobado para enfermedad coronaria establecida a 2026. Barata, accesible, perfil de seguridad conocido.
El dilema de inhibir la inmunidad innata
Todas estas estrategias comparten un riesgo de fondo: NLRP3 e IL-1 tienen funciones defensivas legítimas. Inhibirlos crónicamente tiene consecuencias:
- CANTOS documentó aumento de infecciones fatales con canakinumab — la prueba más clara de que el bloqueo IL-1 global tiene coste.
- La promesa de los inhibidores directos de NLRP3: ser más selectivos que el bloqueo IL-1 global — inhibir solo el inflamasoma NLRP3 (no IL-1 de otras fuentes) podría preservar más función defensiva. Pero esto aún debe demostrarse clínicamente.
- La ventaja de la modulación lifestyle: ayuno (BHB), ejercicio, restricción calórica reducen activación NLRP3 sin abolirla — modulación fisiológica, no bloqueo farmacológico.
El landscape de inhibidores NLRP3 está documentado en detalle en inhibidores y geroenzimas: landscape farmacológico 2026. La compra de Ventyx por Eli Lilly marca el momento en que la clase NLRP3 pasó de promesa académica a apuesta de big pharma — pero a 2026 ningún inhibidor directo de NLRP3 está aún aprobado por la FDA. La única terapia antiinflamatoria aprobada para enfermedad coronaria sigue siendo la colchicina.
NLRP3 es el inflamasoma central del inflammaging y una de las dianas antiinflamatorias del envejecimiento más prometedoras de 2026. La validación clínica de la vía IL-1 (CANTOS, colchicina aprobada FDA 2023) es sólida, pero ningún inhibidor directo de NLRP3 está aprobado aún. La compra de Ventyx por Eli Lilly (1.200 millones, enero 2026) marca el momento de la clase. Mientras tanto, las palancas con mecanismo NLRP3 disponibles hoy son lifestyle (ayuno/BHB, ejercicio) y, en contexto cardiovascular, colchicina.
Este artículo es cobertura editorial del inflamasoma NLRP3 y sus mecanismos de modulación. La biología molecular está basada en literatura primaria (Tschopp et al. 2002 — concepto inflamasoma; Latz & Duewell 2018 Semin Immunol PMID 30268598 — NLRP3 en inflammaging; revisiones en Cell Mol Immunol 2021-2025 sobre regulación y activación; más de 15.000 publicaciones sobre NLRP3). Los datos clínicos están basados en publicaciones revisadas por pares y comunicaciones oficiales hasta mayo 2026 (CANTOS — Ridker et al. NEJM 2017, NCT01327846; LoDoCo2 y COLCOT — colchicina cardiovascular; datos VTX3232 presentados en MDS 2025; comunicado de adquisición Eli Lilly/Ventyx 7 enero 2026). Este texto NO constituye recomendación médica individual ni prescripción. A mayo de 2026 NO existe ningún inhibidor directo de NLRP3 aprobado por FDA o EMA. Los inhibidores directos (MCC950, VTX3232, VTX2735, dapansutrile, NT-0796, selnoflast) están en investigación preclínica o clínica (Phase 1/2). VTX3232 (Ventyx, ahora Eli Lilly) tiene datos Phase 2 en Parkinson precoz (bien tolerado, mejoras en MDS-UPDRS, reducción de IL-1β/IL-18) pero requiere Phase 2 placebo-controlado y Phase 3 antes de cualquier aprobación; los datos provienen de un estudio pequeño (10 pacientes). MCC950 no avanzó clínicamente por hepatotoxicidad. Los inhibidores downstream anti-IL-1 (canakinumab/Ilaris, anakinra/Kineret, rilonacept/Arcalyst) están aprobados FDA para indicaciones específicas (CAPS, gota refractaria, artritis, pericarditis) pero NO para enfermedad cardiovascular: canakinumab demostró beneficio CV en CANTOS pero NO obtuvo aprobación CV y aumentó el riesgo de infección fatal. La colchicina low-dose (Lodoco) SÍ está aprobada por FDA (2023) para reducción de riesgo cardiovascular en enfermedad coronaria establecida — pero su uso es decisión clínica individual con cardiología (tiene interacciones farmacológicas y efectos gastrointestinales). El disulfiram está aprobado solo para alcoholismo; su uso para condiciones inflamatorias es experimental (off-label/repurposing). Las intervenciones lifestyle discutidas (ayuno intermitente, restricción calórica, ejercicio, dieta cetogénica que eleva BHB) tienen mecanismo molecular plausible sobre NLRP3 pero su magnitud de efecto antiinflamatorio en humanos es modesta y variable. Los datos cuantitativos (reducción ~15% MACE en CANTOS, ~23% en LoDoCo2, valor ~1.200 millones de la compra Ventyx) son cifras específicas de sus contextos. Inhibir la inmunidad innata crónicamente conlleva riesgo de inmunosupresión e infección — NLRP3 e IL-1 tienen funciones defensivas legítimas. Los lectores con enfermedades inflamatorias (gota, CAPS, pericarditis), cardiovasculares (enfermedad coronaria, post-infarto), neurodegenerativas (Parkinson, Alzheimer) o metabólicas que consideren terapias antiinflamatorias deben consultar con reumatología, cardiología, neurología o medicina interna habilitada. La medicina antiinflamatoria del envejecimiento es campo en evolución acelerada — KRECE actualizará esta entrada cuando algún inhibidor directo de NLRP3 reciba aprobación regulatoria, cuando VTX3232 publique Phase 2 placebo-controlado, o cuando emerjan datos relevantes sobre la combinación senolíticos + inhibidores NLRP3.
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417-426. (Acuñación del concepto inflamasoma)
- Latz E, Duewell P. NLRP3 inflammasome activation in inflammaging. Semin Immunol. 2018;40:61-73. PMID 30268598. (NLRP3 en inflammaging y metaflammation)
- Swanson KV, Deng M, Ting JP. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol Immunol. 2021;18:1141-1160. (Mecanismos de regulación)
- Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease (CANTOS). N Engl J Med. 2017;377:1119-1131. NCT01327846. (Validación hipótesis inflamatoria CV)
- Nidorf SM, Fiolet ATL, Mosterd A, et al. Colchicine in Patients with Chronic Coronary Disease (LoDoCo2). N Engl J Med. 2020;383:1838-1847. (Colchicina cardiovascular)
- Tardif JC, Kouz S, Waters DD, et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction (COLCOT). N Engl J Med. 2019;381:2497-2505.
- Eli Lilly and Company. Lilly to acquire Ventyx Biosciences to advance oral therapies targeting inflammatory-mediated diseases. PR Newswire. 7 enero 2026. (Adquisición Ventyx ~1.200M, plataforma NLRP3)
- Gregg R, et al. NLRP3 Inhibitor VTX3232 Well Tolerated, Improves Parkinson Symptoms in Phase 2a Trial. Presentado en MDS 2025, Honolulu. (VTX3232 Parkinson)
- He WT, Wan H, Hu L, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25:1285-1298. (Gasdermina D y piroptosis)
- Coll RC, Robertson AAB, Chae JJ, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases (MCC950). Nat Med. 2015;21:248-255. (MCC950, inhibidor prototipo)
- Youm YH, Nguyen KY, Grant RW, et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. 2015;21:263-269. (BHB inhibe NLRP3)
- Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357-1361. (Cristales de colesterol y aterosclerosis)
- Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237-241. (Gota y NLRP3)
- Halle A, Hornung V, Petzold GC, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat Immunol. 2008;9:857-865. (β-amiloide y Alzheimer)
- Venegas C, Kumar S, Franklin BS, et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature. 2017;552:355-361. (ASC specks siembran amiloide)
- Marchetti C, Swartzwelter B, Gamboni F, et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome (dapansutrile). PNAS. 2018;115:E1530-E1539.
- Fierce Biotech. Lilly buys inflammation biotech Ventyx for $1.2B in wake of Parkinson’s, CV readouts. 8 enero 2026. (Detalles de la operación y pipeline Ventyx)
- He Y, Zeng MY, Yang D, Motro B, Núñez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530:354-357. (NEK7 licenciador)